Fan Caoqi, Cahoon Jordan L, Dinh Bryan L, Ortega-Del Vecchyo Diego, Huber Christian, Edge Michael D, Mancuso Nicholas, Chiang Charleston W K
Department of Quantitative and Computational Biology, University of Southern California.
Center for Genetic Epidemiology, Department of Population and Public Health Sciences, Keck School of Medicine, University of Southern California.
bioRxiv. 2023 Oct 13:2023.10.10.561787. doi: 10.1101/2023.10.10.561787.
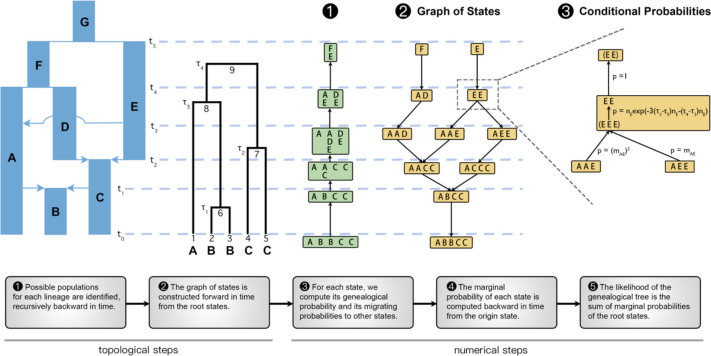
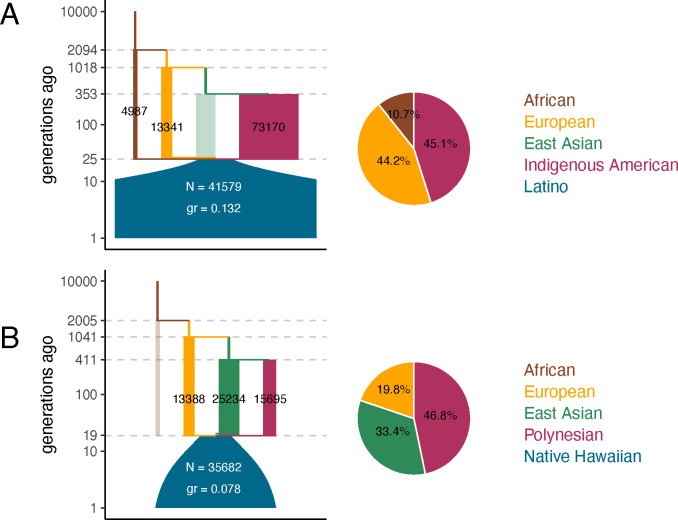
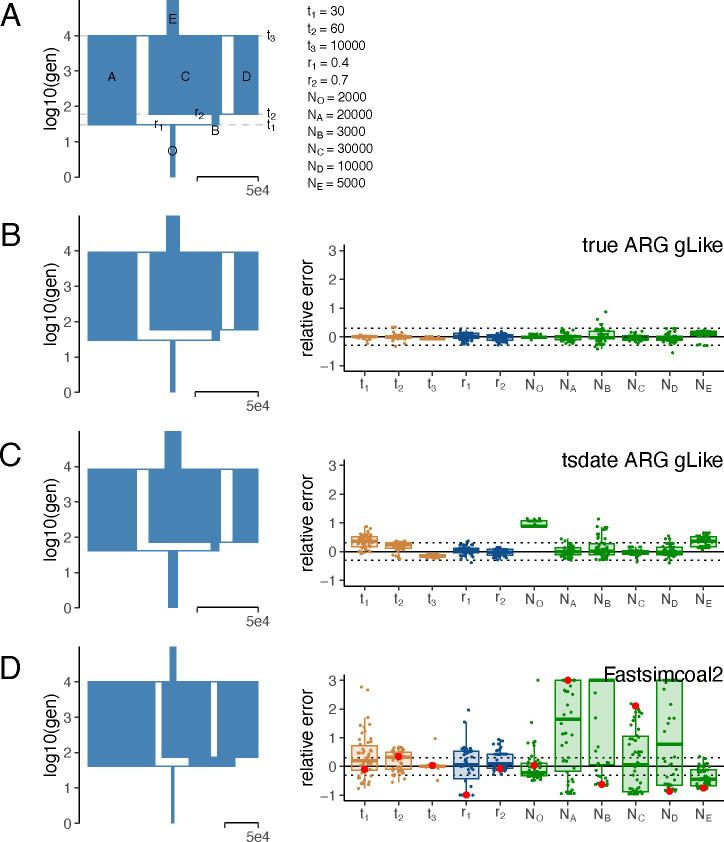
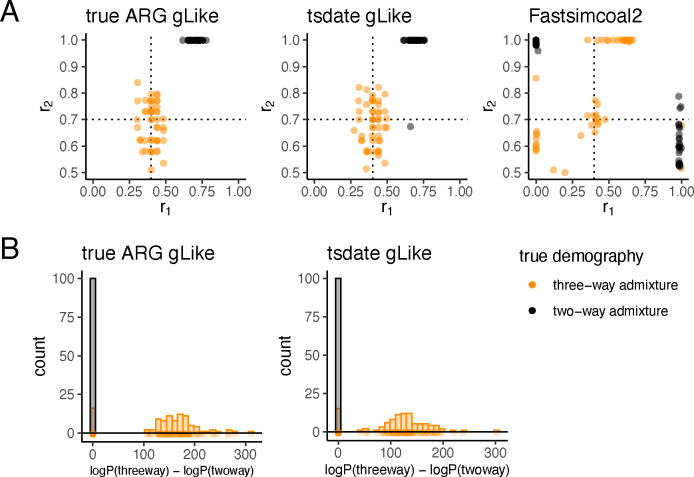
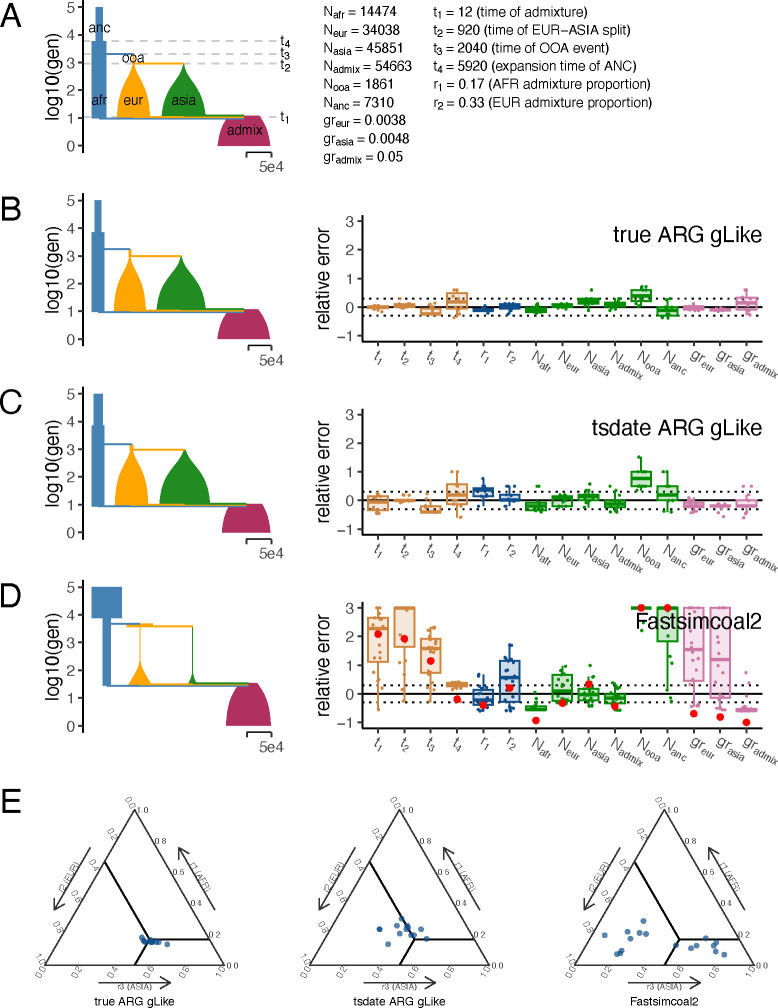
The demographic history of a population drives the pattern of genetic variation and is encoded in the gene-genealogical trees of the sampled alleles. However, existing methods to infer demographic history from genetic data tend to use relatively low-dimensional summaries of the genealogy, such as allele frequency spectra. As a step toward capturing more of the information encoded in the genome-wide sequence of genealogical trees, here we propose a novel framework called the genealogical likelihood (gLike), which derives the full likelihood of a genealogical tree under any hypothesized demographic history. Employing a graph-based structure, gLike summarizes across independent trees the relationships among all lineages in a tree with all possible trajectories of population memberships through time and efficiently computes the exact marginal probability under a parameterized demographic model. Through extensive simulations and empirical applications on populations that have experienced multiple admixtures, we showed that gLike can accurately estimate dozens of demographic parameters when the true genealogy is known, including ancestral population sizes, admixture timing, and admixture proportions. Moreover, when using genealogical trees inferred from genetic data, we showed that gLike outperformed conventional demographic inference methods that leverage only the allele-frequency spectrum and yielded parameter estimates that align with established historical knowledge of the past demographic histories for populations like Latino Americans and Native Hawaiians. Furthermore, our framework can trace ancestral histories by analyzing a sample from the admixed population without proxies for its source populations, removing the need to sample ancestral populations that may no longer exist. Taken together, our proposed gLike framework harnesses underutilized genealogical information to offer exceptional sensitivity and accuracy in inferring complex demographies for humans and other species, particularly as estimation of genome-wide genealogies improves.
一个种群的人口统计学历史驱动着遗传变异模式,并编码在抽样等位基因的基因系谱树中。然而,现有的从遗传数据推断人口统计学历史的方法往往使用基因系谱的相对低维摘要,例如等位基因频率谱。作为朝着捕捉基因系谱树全基因组序列中更多编码信息迈出的一步,我们在此提出一种名为系谱似然(gLike)的新框架,它能推导出在任何假设的人口统计学历史下基因系谱树的全似然。利用基于图的结构,gLike总结了独立树中一棵树内所有谱系之间的关系以及种群成员随时间的所有可能轨迹,并在参数化人口模型下高效计算精确的边际概率。通过对经历多次混合的种群进行广泛模拟和实证应用,我们表明当真实系谱已知时,gLike可以准确估计几十个人口统计学参数,包括祖先种群大小、混合时间和混合比例。此外,当使用从遗传数据推断出的基因系谱树时,我们表明gLike优于仅利用等位基因频率谱的传统人口统计学推断方法,并产生与拉丁裔美国人和夏威夷原住民等种群过去人口统计学历史的既定历史知识相符的参数估计。此外,我们的框架可以通过分析混合种群的样本追溯祖先历史,而无需其源种群的代理,从而无需对可能不再存在的祖先种群进行抽样。综上所述,我们提出的gLike框架利用了未充分利用的系谱信息,在推断人类和其他物种的复杂人口统计学时提供了卓越的敏感性和准确性,特别是随着全基因组系谱估计的改进。