Yoo Pilsun, Bhowmik Debsindhu, Mehta Kshitij, Zhang Pei, Liu Frank, Lupo Pasini Massimiliano, Irle Stephan
Computational Sciences and Engineering Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN, 37831, USA.
Computer Science and Mathematics Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN, 37831, USA.
Sci Rep. 2023 Nov 16;13(1):20031. doi: 10.1038/s41598-023-45385-9.
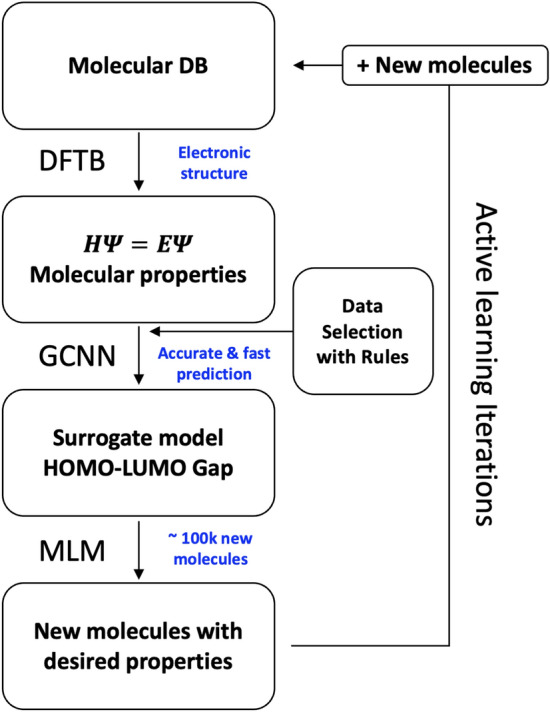
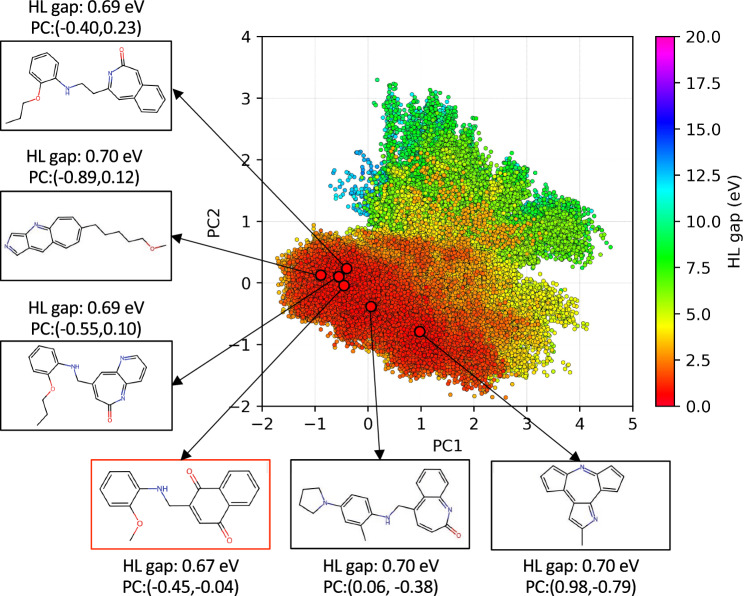
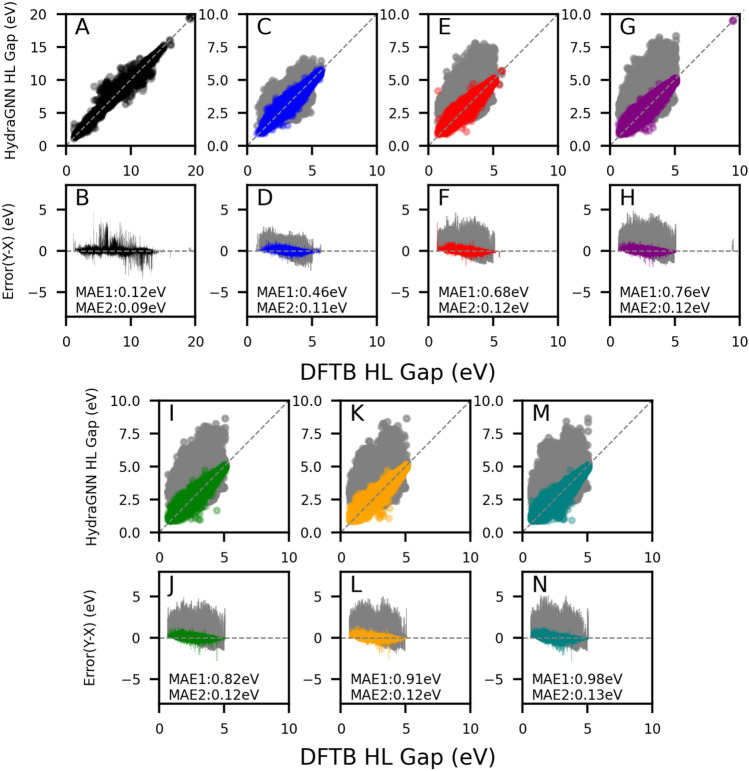
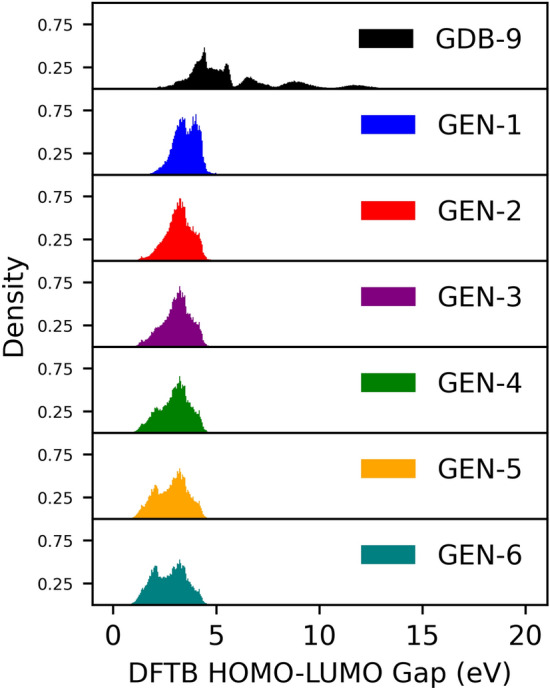
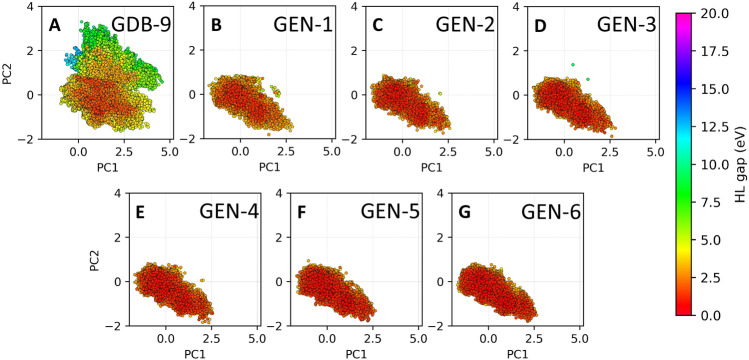
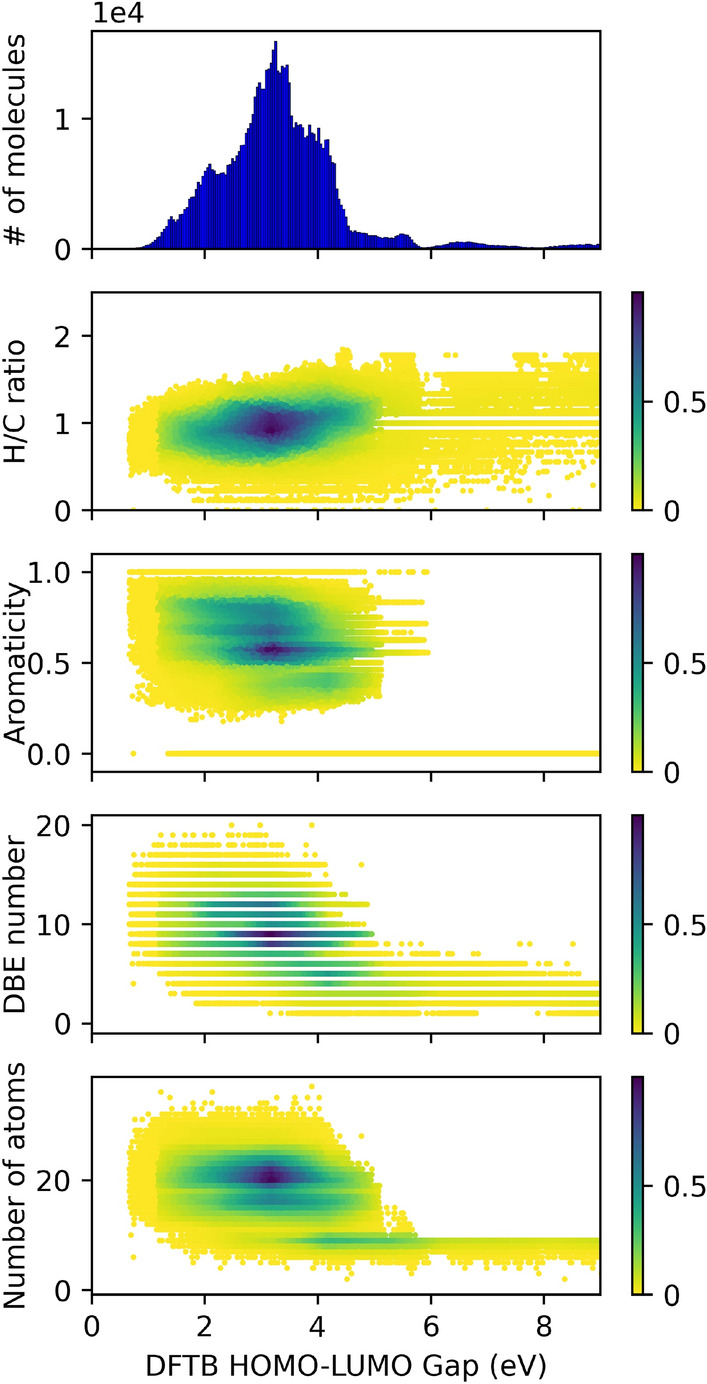
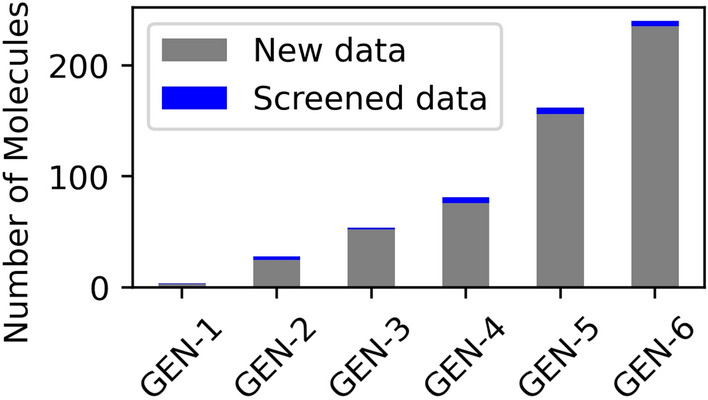
The inverse design of novel molecules with a desirable optoelectronic property requires consideration of the vast chemical spaces associated with varying chemical composition and molecular size. First principles-based property predictions have become increasingly helpful for assisting the selection of promising candidate chemical species for subsequent experimental validation. However, a brute-force computational screening of the entire chemical space is decidedly impossible. To alleviate the computational burden and accelerate rational molecular design, we here present an iterative deep learning workflow that combines (i) the density-functional tight-binding method for dynamic generation of property training data, (ii) a graph convolutional neural network surrogate model for rapid and reliable predictions of chemical and physical properties, and (iii) a masked language model. As proof of principle, we employ our workflow in the iterative generation of novel molecules with a target energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).
设计具有理想光电特性的新型分子需要考虑与化学成分和分子大小变化相关的巨大化学空间。基于第一性原理的性质预测对于辅助选择有前景的候选化学物种以进行后续实验验证越来越有帮助。然而,对整个化学空间进行蛮力计算筛选显然是不可能的。为了减轻计算负担并加速合理的分子设计,我们在此提出一种迭代深度学习工作流程,该流程结合了:(i) 用于动态生成性质训练数据的密度泛函紧束缚方法;(ii) 用于快速可靠预测化学和物理性质的图卷积神经网络替代模型;以及 (iii) 一种掩码语言模型。作为原理证明,我们将我们的工作流程用于迭代生成在最高占据分子轨道 (HOMO) 和最低未占据分子轨道 (LUMO) 之间具有目标能隙的新型分子。