Arfeuille Chloé, Vial Yoann, Cadenet Margaux, Caye-Eude Aurélie, Fenneteau Odile, Neven Quentin, Bonnard Adeline A, Pizzi Simone, Carpentieri Giovanna, Capri Yline, Girardi Katia, Pedace Lucia, Macchiaiolo Marina, Boudhar Kamel, Khaled Monia Ben, Chahla Wadih Abou, Lutun Anne, Fahd Mony, Drunat Séverine, Flex Elisabetta, Dalle Jean-Hugues, Strullu Marion, Locatelli Franco, Tartaglia Marco, Cavé Hélène
Département de Génétique, Unité de Génétique Moléculaire, Hôpital Robert Debré, Assistance Publique des Hôpitaux de Paris (AP-HP), Paris, France; INSERM UMR_S1131, Institut de Recherche Saint-Louis, Université Paris-Cité, Paris.
Service d'Hématologie Biologique, Hôpital Robert Debré, Assistance Publique des Hôpitaux de Paris (AP-HP), Paris.
Haematologica. 2024 Aug 1;109(8):2542-2554. doi: 10.3324/haematol.2023.283917.
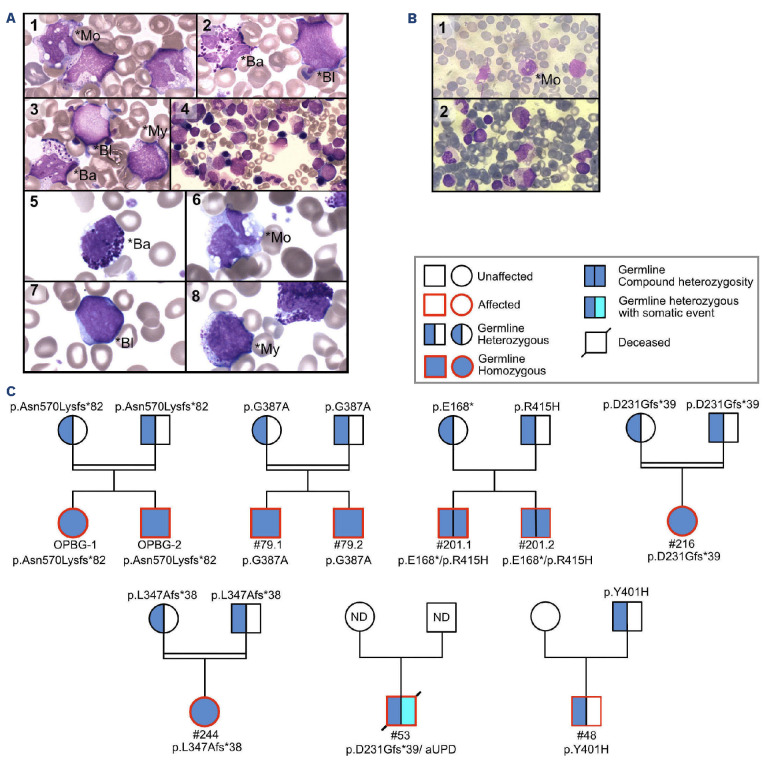
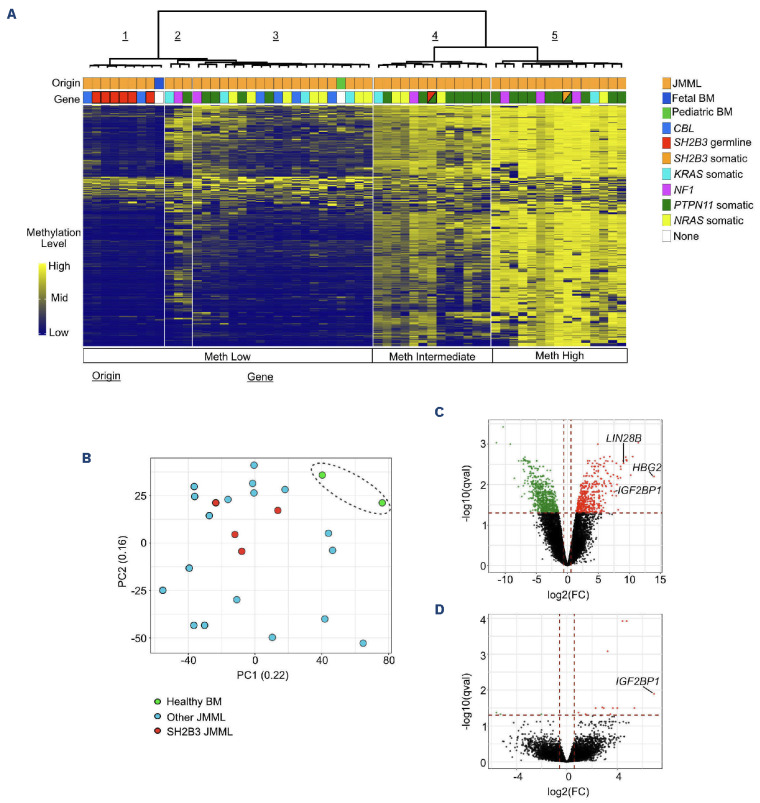
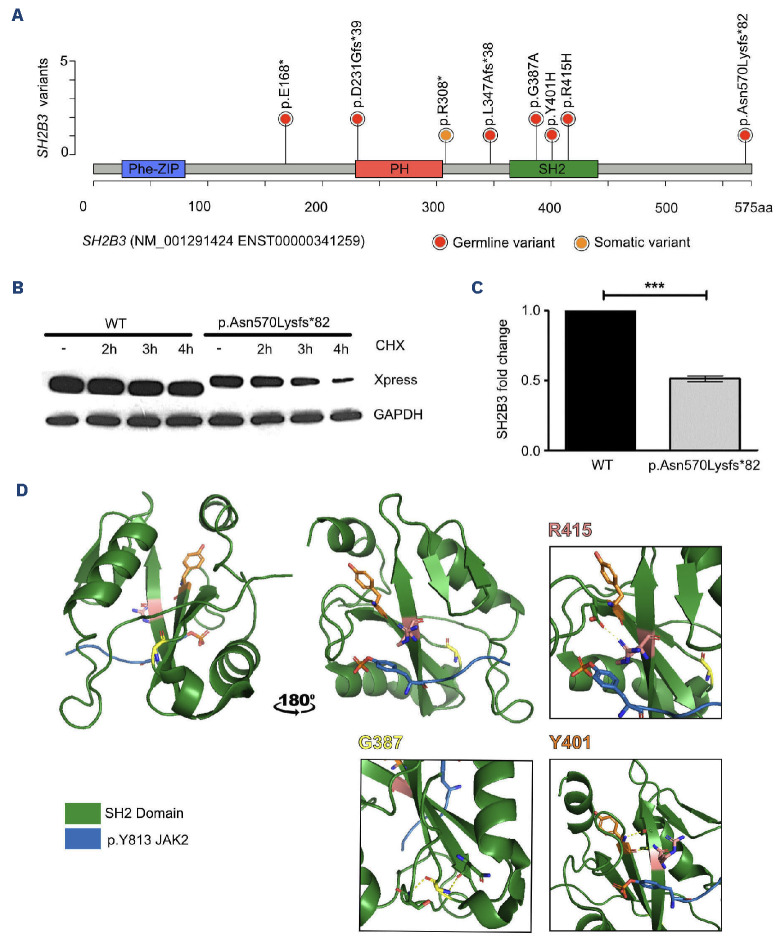
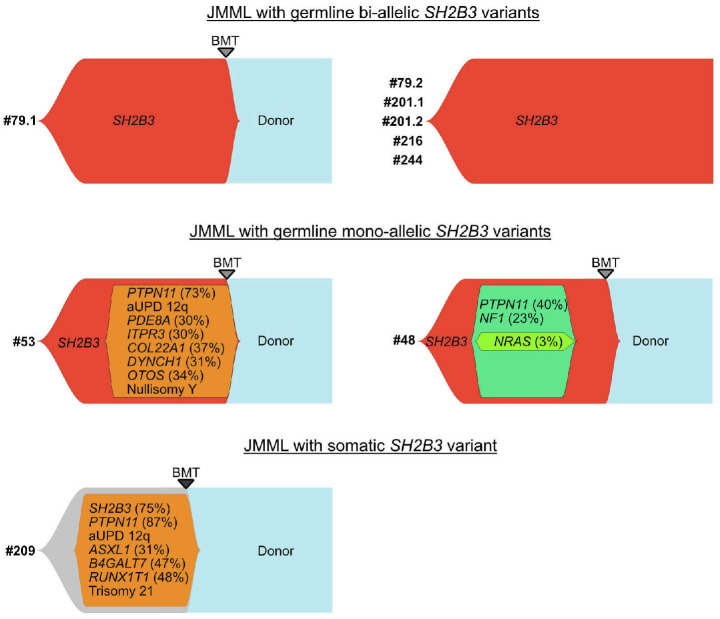
Juvenile myelomonocytic leukemia (JMML) is a rare, generally aggressive myeloproliferative neoplasm affecting young children. It is characterized by granulomonocytic expansion, with monocytosis infiltrating peripheral tissues. JMML is initiated by mutations upregulating RAS signaling. Approximately 10% of cases remain without an identified driver event. Exome sequencing of two unrelated cases of familial JMML of unknown genetics and analysis of the French JMML cohort identified 11 patients with variants in SH2B3, encoding LNK, a negative regulator of the JAK-STAT pathway. All variants were absent from healthy population databases, and the mutation spectrum was consistent with a loss of function of the LNK protein. A stoploss variant was shown to affect both protein synthesis and stability. The other variants were either truncating or missense, the latter affecting the SH2 domain that interacts with activated JAK. Of the 11 patients, eight from five families inherited pathogenic bi-allelic SH2B3 germline variants from their unaffected heterozygous parents. These children represent half of the cases with no identified causal mutation in the French cohort. They displayed typical clinical and hematologic features of JMML with neonatal onset and marked thrombocytopenia. They had a hypomethylated DNA profile with fetal characteristics and did not have additional genetic alterations. All patients showed partial or complete spontaneous clinical resolution. However, progression to thrombocythemia and immunity-related pathologies may be of concern later in life. Bi-allelic SH2B3 germline mutations thus define a new condition predisposing to a JMML-like disorder, suggesting that JAK pathway deregulation is capable of initiating JMML, and opening new therapeutic options.
青少年骨髓单核细胞白血病(JMML)是一种罕见的、通常具有侵袭性的骨髓增殖性肿瘤,影响幼儿。其特征是粒单核细胞扩增,单核细胞增多浸润外周组织。JMML由上调RAS信号的突变引发。约10%的病例未发现驱动事件。对两例无已知遗传学信息的家族性JMML无关病例进行外显子组测序,并对法国JMML队列进行分析,确定了11例SH2B3基因变异的患者,该基因编码LNK,是JAK-STAT途径的负调节因子。所有变异在健康人群数据库中均未出现,且突变谱与LNK蛋白功能丧失一致。一个阻止损失变异被证明影响蛋白质合成和稳定性。其他变异要么是截断变异,要么是错义变异,后者影响与活化JAK相互作用的SH2结构域。在这11例患者中,来自五个家庭的8例从其未受影响的杂合子父母那里遗传了致病性双等位基因SH2B3种系变异。这些儿童占法国队列中未发现因果突变病例的一半。他们表现出JMML典型的临床和血液学特征,新生儿期发病,血小板显著减少。他们具有胎儿特征的低甲基化DNA谱,且没有其他基因改变。所有患者均表现出部分或完全自发的临床缓解。然而,后期可能会发展为血小板增多症和免疫相关疾病。因此,双等位基因SH2B3种系突变定义了一种易患JMML样疾病的新情况,提示JAK途径失调能够引发JMML,并开辟了新的治疗选择。