Radyk Megan D, Nelson Barbara S, Halbrook Christopher J, Wood Alexander, Lavoie Brooke, Salvatore Lucie, Corfas Gabriel, Colacino Justin A, Shah Yatrik M, Crawford Howard C, Lyssiotis Costas A
Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI, USA.
Doctoral Program in Cancer Biology, University of Michigan, Ann Arbor, USA.
bioRxiv. 2023 Nov 8:2023.11.06.565895. doi: 10.1101/2023.11.06.565895.
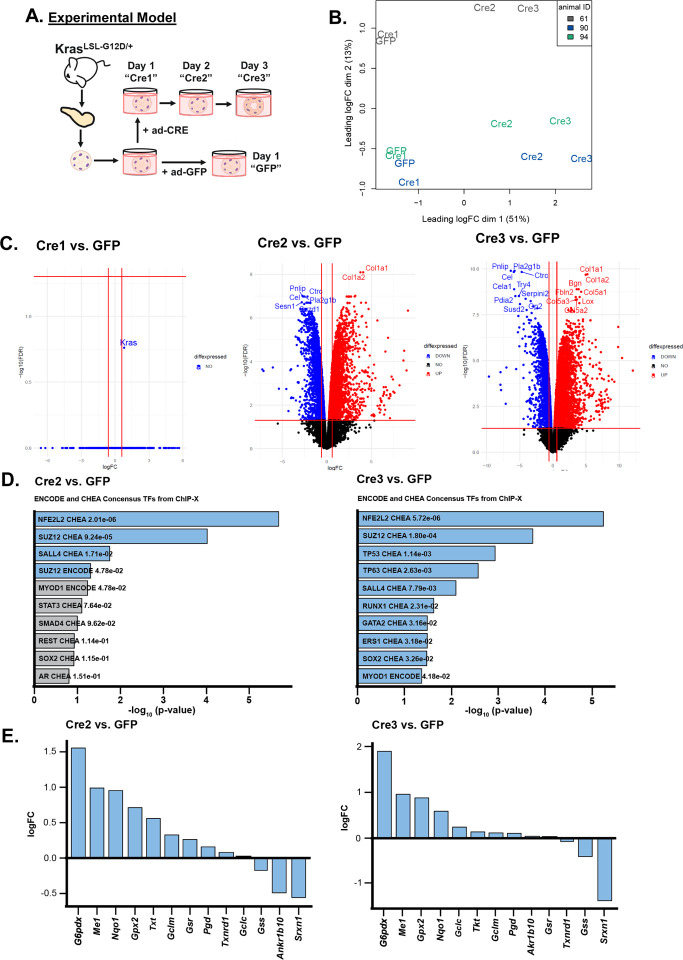
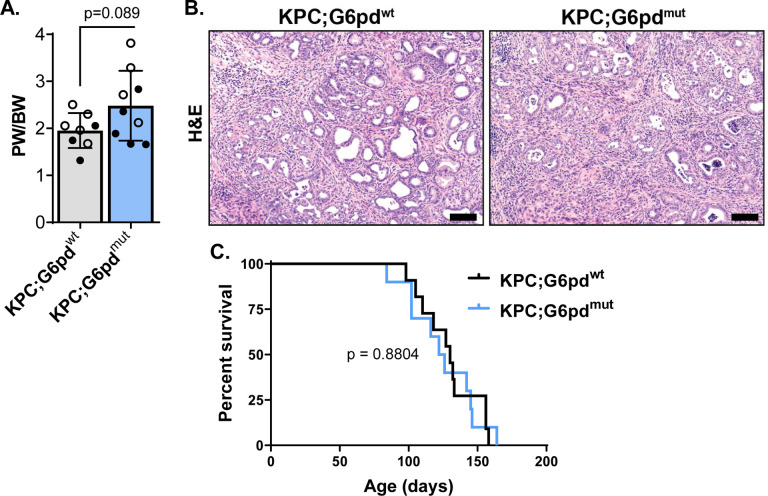
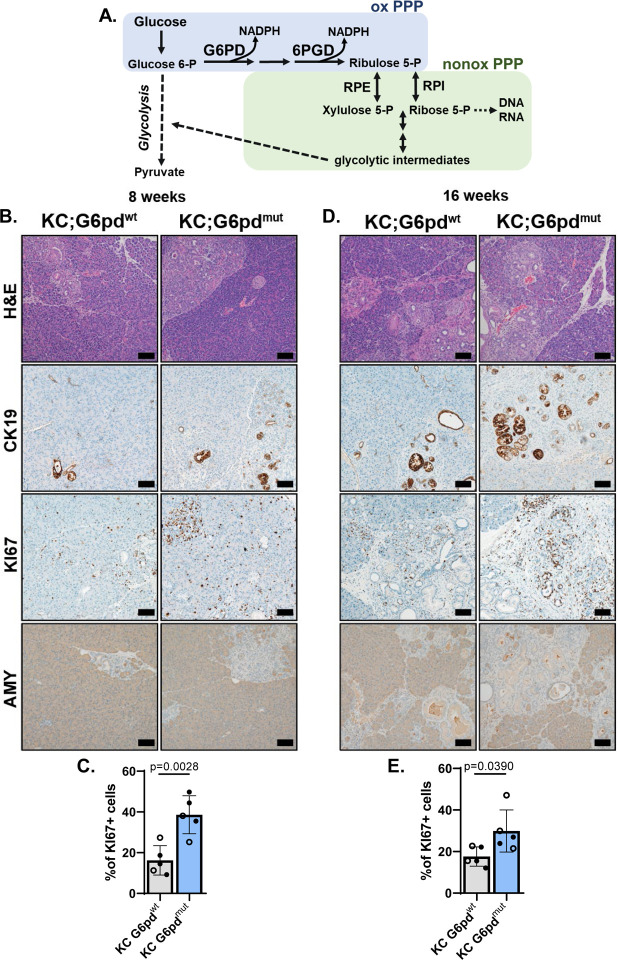
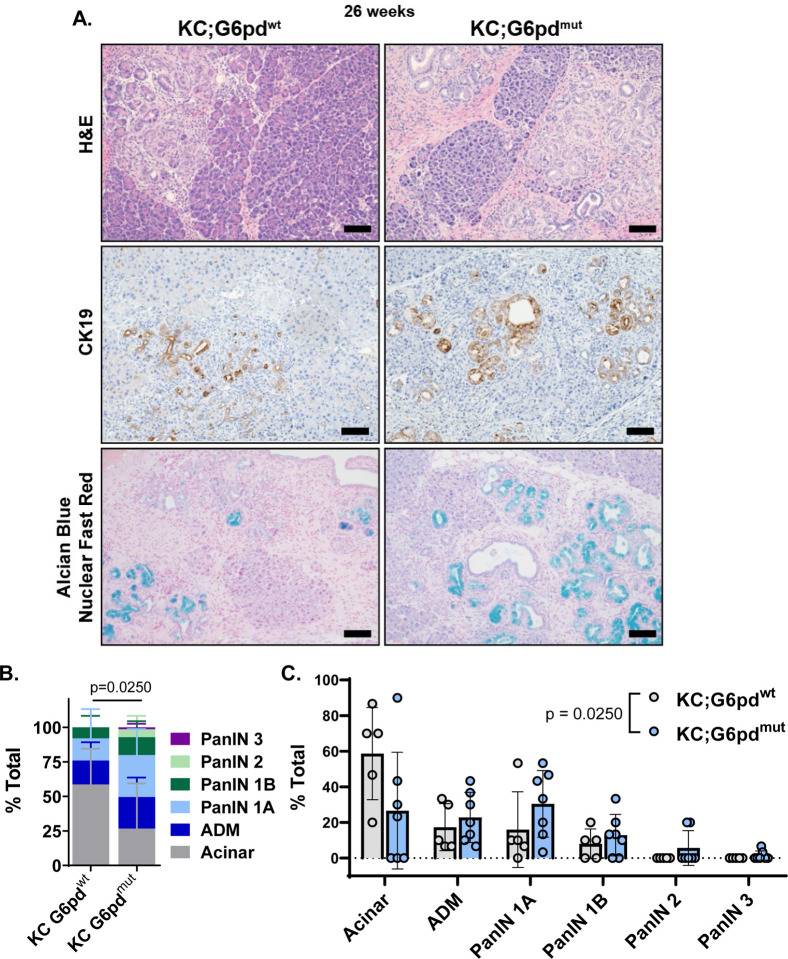
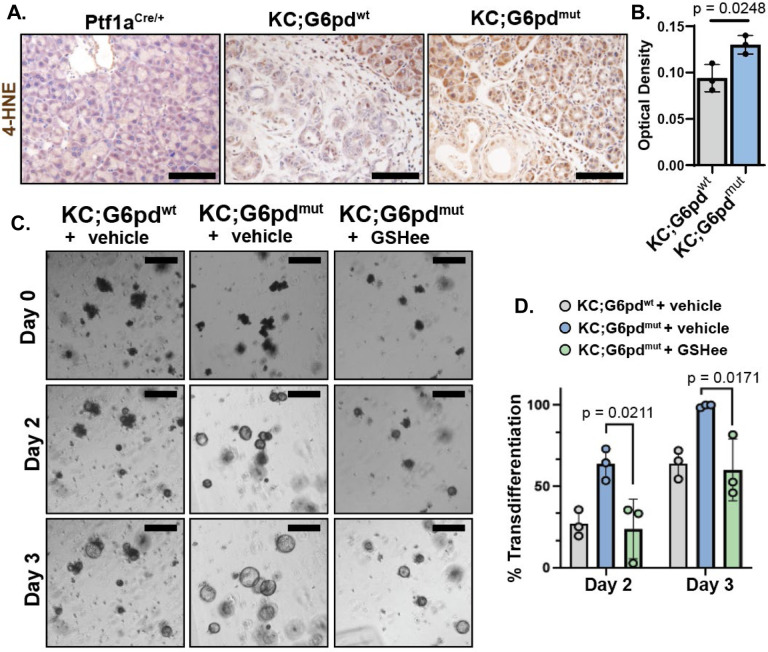
Activating mutations in extensively reprogram cellular metabolism to support the continuous growth, proliferation, and survival of pancreatic tumors. Targeting these metabolic dependencies are promising approaches for the treatment of established tumors. However, metabolic reprogramming is required early during tumorigenesis to provide transformed cells selective advantage towards malignancy. Acinar cells can give rise to pancreatic tumors through acinar-to-ductal metaplasia (ADM). Dysregulation of pathways that maintain acinar homeostasis accelerate tumorigenesis. During ADM, acinar cells transdifferentiate to duct-like cells, a process driven by oncogenic . The metabolic reprogramming that is required for the transdifferentiation in ADM is unclear. We performed transcriptomic analysis on mouse acinar cells undergoing ADM and found metabolic programs are globally enhanced, consistent with the transition of a specialized cell to a less differentiated phenotype with proliferative potential. Indeed, we and others have demonstrated how inhibiting metabolic pathways necessary for ADM can prevent transdifferentiation and tumorigenesis. Here, we also find NRF2-target genes are differentially expressed during ADM. Among these, we focused on the increase in the gene coding for NADPH-producing enzyme, Glucose-6-phosphate dehydrogenase (G6PD). Using established mouse models of -driven pancreatic tumorigenesis and G6PD-deficiency, we find that mutant accelerates ADM and pancreatic intraepithelial neoplasia. Acceleration of cancer initiation with G6PD-deficiency is dependent on its NADPH-generating function in reactive oxygen species (ROS) management, as opposed to other outputs of the pentose phosphate pathway. Together, this work provides new insights into the function of metabolic pathways during early tumorigenesis.
激活突变会广泛地重新编程细胞代谢,以支持胰腺肿瘤的持续生长、增殖和存活。针对这些代谢依赖性是治疗已形成肿瘤的有前景的方法。然而,在肿瘤发生早期就需要代谢重编程,以赋予转化细胞对恶性肿瘤的选择性优势。腺泡细胞可通过腺泡-导管化生(ADM)产生胰腺肿瘤。维持腺泡内环境稳定的信号通路失调会加速肿瘤发生。在ADM过程中,腺泡细胞转分化为导管样细胞,这一过程由致癌作用驱动。ADM中转分化所需的代谢重编程尚不清楚。我们对经历ADM的小鼠腺泡细胞进行了转录组分析,发现代谢程序整体增强,这与一个特化细胞向具有增殖潜力的低分化表型的转变一致。事实上,我们和其他人已经证明了抑制ADM所需的代谢途径如何能够阻止转分化和肿瘤发生。在这里,我们还发现NRF2靶基因在ADM过程中差异表达。其中,我们重点关注了编码产生NADPH的酶葡萄糖-6-磷酸脱氢酶(G6PD)的基因的增加。利用已建立的由驱动的胰腺肿瘤发生和G6PD缺陷的小鼠模型,我们发现突变体加速了ADM和胰腺上皮内瘤变。G6PD缺陷导致癌症起始加速依赖于其在活性氧(ROS)管理中的NADPH生成功能,而不是磷酸戊糖途径的其他产物。总之,这项工作为肿瘤发生早期代谢途径的功能提供了新的见解。