Burleigh Alice, Omoyinmi Ebun, Papadopoulou Charalampia, Al-Abadi Eslam, Hong Ying, Price-Kuehne Fiona, Moraitis Elena, Titheradge Hannah, Montesi Francesca, Xu Diane, Eleftheriou Despina, Brogan Paul
Infection, Immunity and Inflammation, University College London Great Ormond Street Institute of Child Health, London, UK.
Centre for Adolescent Rheumatology Versus Arthritis at University College London, London, UK.
Rheumatology (Oxford). 2024 Dec 1;63(12):3457-3470. doi: 10.1093/rheumatology/kead628.
Several monogenic autoinflammatory disorders and primary immunodeficiencies can present early in life with features that may be mistaken for Behçet's disease (BD). We aimed to develop a genetic analysis workflow to identify rare monogenic BD-like diseases and establish the contribution of HLA haplotype in a cohort of patients from the UK.
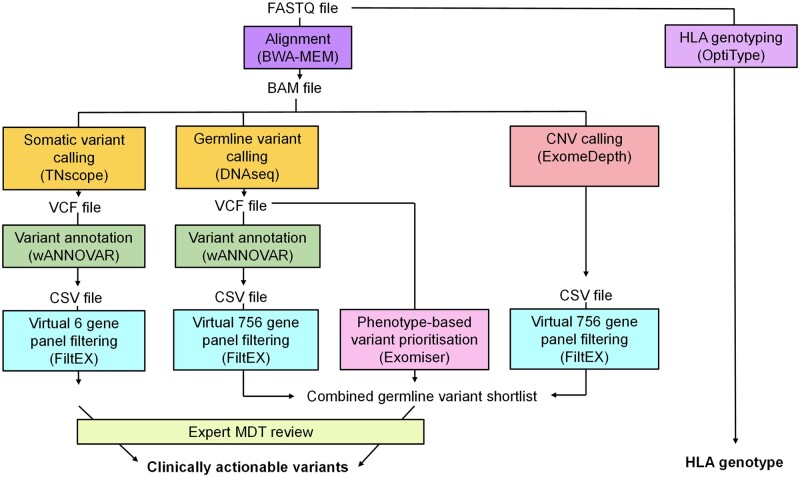
Patients with clinically suspected BD were recruited from four BD specialist care centres in the UK. All participants underwent whole-exome sequencing (WES), and genetic analysis thereafter by (i) examining genes known to cause monogenic immunodeficiency, autoinflammation or vasculitis by virtual panel application; (ii) scrutiny of variants prioritized by Exomiser using Human Phenotype Ontology (HPO); (iii) identification of copy number variants using ExomeDepth; and (iv) HLA-typing using OptiType.
Thirty-one patients were recruited: median age 15 (4-52), and median disease onset age 5 (0-20). Nine/31 (29%) patients had monogenic disease mimicking BD: five cases of Haploinsufficiency of A20 with novel TNFAIP3 variants (p.T76I, p. M112Tfs8, p. S548Dfs128, p. C657Vfs14, p. E661Nfs36); one case of ISG15 deficiency with a novel nonsense variant (ISG15: p.Q16X) and 1p36.33 microdeletion; one case of common variable immune deficiency (TNFRSF13B: p.A181E); and two cases of TNF receptor-associated periodic syndrome (TNFRSF1A: p.R92Q). Of the remaining 22 patients, eight (36%) were HLA-B*51 positive.
We describe a novel genetic workflow for BD, which can efficiently detect known and potentially novel monogenic forms of BD, whilst additionally providing HLA-typing. Our results highlight the importance of genetic testing before BD diagnosis, as this has an impact on choice of therapy, prognosis and genetic counselling.
几种单基因自身炎症性疾病和原发性免疫缺陷在生命早期可能表现出与白塞病(BD)相似的特征,易被误诊。我们旨在开发一种基因分析流程,以识别罕见的单基因BD样疾病,并确定HLA单倍型在一组英国患者中的作用。
从英国四个BD专科护理中心招募临床疑似BD患者。所有参与者均接受全外显子组测序(WES),随后进行基因分析,方法包括:(i)通过虚拟面板应用检查已知会导致单基因免疫缺陷、自身炎症或血管炎的基因;(ii)使用人类表型本体论(HPO)对Exomiser优先排序的变异进行仔细审查;(iii)使用ExomeDepth识别拷贝数变异;(iv)使用OptiType进行HLA分型。
共招募31例患者,中位年龄15岁(4 - 52岁),疾病中位发病年龄5岁(0 - 20岁)。31例患者中有9例(29%)患有模仿BD的单基因疾病:5例A20单倍体不足伴新型TNFAIP3变异(p.T76I、p.M112Tfs8、p.S548Dfs128、p.C657Vfs14、p.E661Nfs36);1例ISG15缺陷伴新型无义变异(ISG15:p.Q16X)和1p36.33微缺失;1例常见可变免疫缺陷(TNFRSF13B:p.A181E);2例TNF受体相关周期性综合征(TNFRSF1A:p.R92Q)。其余22例患者中,8例(36%)HLA - B*51阳性。
我们描述了一种针对BD的新型基因分析流程,该流程可有效检测已知的和潜在的新型单基因BD形式,同时还能进行HLA分型。我们的结果强调了在BD诊断前进行基因检测的重要性,因为这会影响治疗选择、预后和遗传咨询。