School of Public Health and the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA.
Genome Med. 2023 Nov 28;15(1):101. doi: 10.1186/s13073-023-01253-9.
Common and rare variants contribute to the etiology of complex traits. However, the extent to which the phenotypic effects of common and rare variants involve shared molecular mediators remains poorly understood. The question is essential to the basic and translational goals of the science of genomics, with critical basic-science, methodological, and clinical consequences.
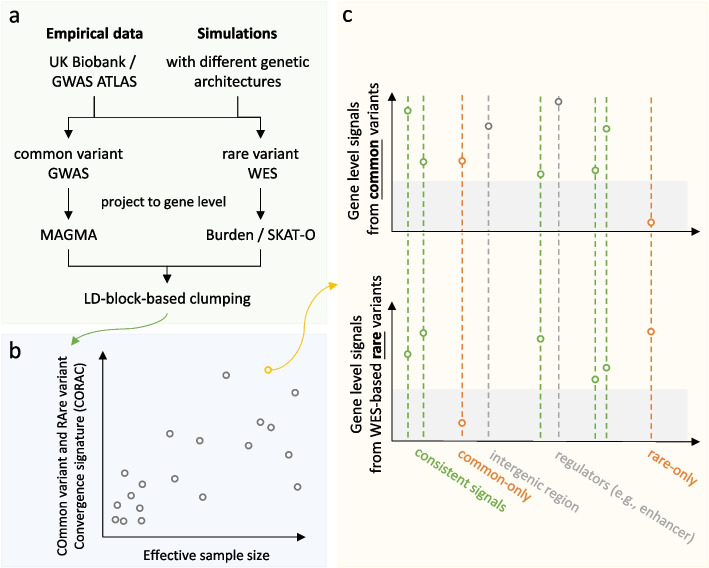
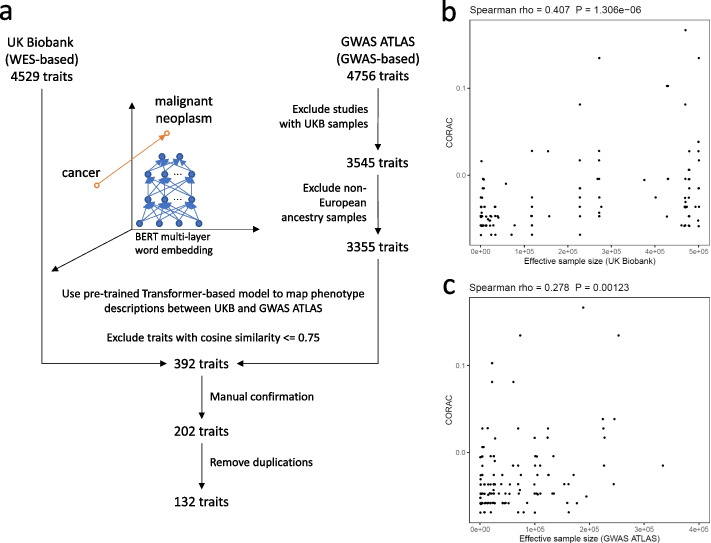
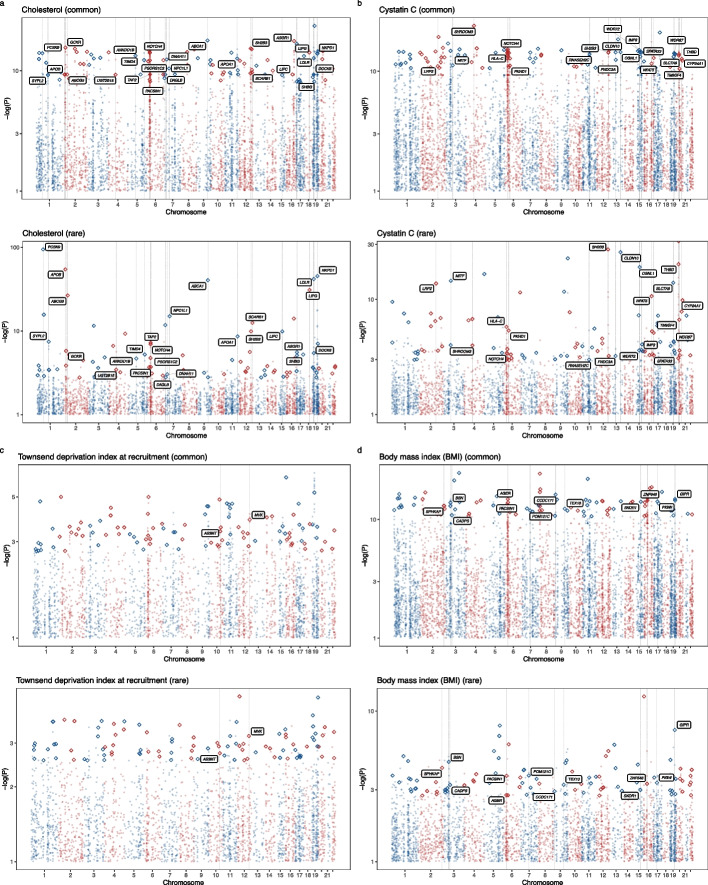
Leveraging the latest release of whole-exome sequencing (WES, for rare variants) and genome-wide association study (GWAS, for common variants) data from the UK Biobank, we developed a metric, the COmmon variant and RAre variant Convergence (CORAC) signature, to quantify the convergence for a broad range of complex traits. We characterized the relationship between CORAC and effective sample size across phenome-wide association studies.
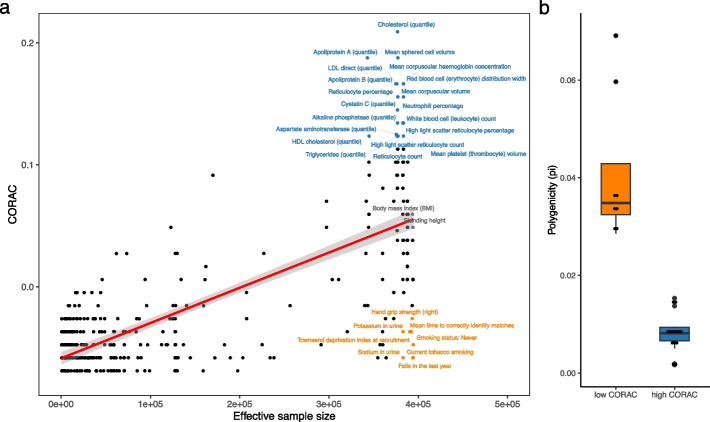
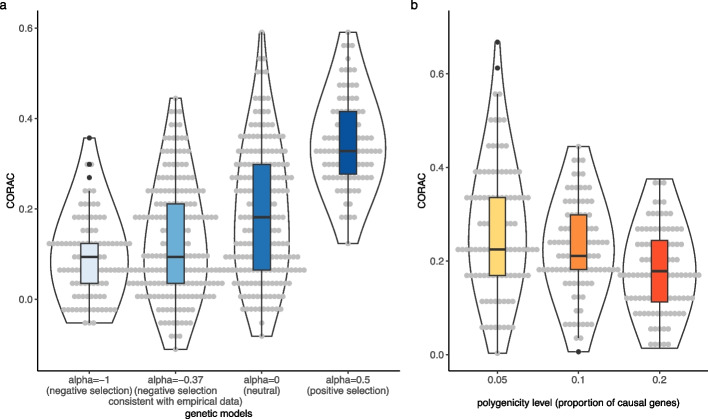
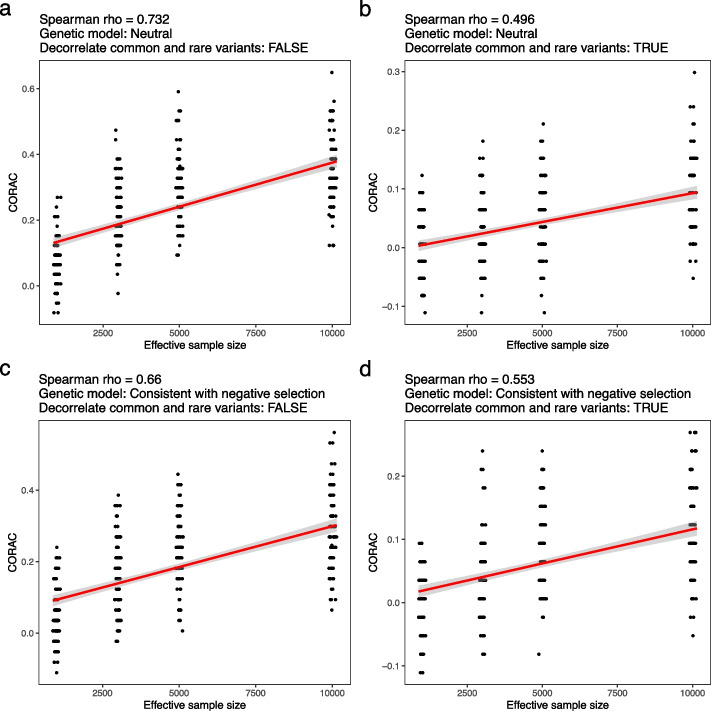
We found that the signature is positively correlated with effective sample size (Spearman ρ = 0.594, P < 2.2e - 16), indicating increased functional convergence of trait-associated genetic variation, across the allele frequency spectrum, with increased power. Sensitivity analyses, including accounting for heteroskedasticity and varying the number of detected association signals, further strengthened the validity of the finding. In addition, consistent with empirical data, extensive simulations showed that negative selection, in line with enhancing polygenicity, has a dampening effect on the convergence signature. Methodologically, leveraging the convergence leads to enhanced association analysis.
The presented framework for the convergence signature has important implications for fine-mapping strategies and drug discovery efforts. In addition, our study provides a blueprint for the expectation from future large-scale whole-genome sequencing (WGS)/WES and sheds methodological light on post-GWAS studies.
常见变异和稀有变异都对复杂性状的病因有影响。然而,常见变异和稀有变异的表型效应在多大程度上涉及共同的分子介质仍知之甚少。这个问题对于基因组学的基础和转化目标至关重要,具有关键的基础科学、方法学和临床意义。
利用英国生物库(UK Biobank)最新发布的全外显子组测序(WES,用于稀有变异)和全基因组关联研究(GWAS,用于常见变异)数据,我们开发了一种度量标准,即常见变异和稀有变异收敛(CORAC)特征,用于量化广泛的复杂性状的收敛程度。我们描述了 CORAC 与全表型关联研究中的有效样本量之间的关系。
我们发现该特征与有效样本量呈正相关(Spearman ρ=0.594,P<2.2e-16),表明随着功效的增加,与性状相关的遗传变异在整个等位基因频率谱上的功能收敛性增加。包括考虑异方差和改变检测到的关联信号数量的敏感性分析进一步加强了这一发现的有效性。此外,与经验数据一致,广泛的模拟表明,负选择与增强多效性一致,对收敛特征具有抑制作用。从方法学上讲,利用收敛性可以增强关联分析。
本文提出的收敛特征框架对精细映射策略和药物发现工作具有重要意义。此外,我们的研究为未来大规模全基因组测序(WGS)/WES 提供了蓝图,并为 GWAS 后研究提供了方法学上的启示。