Northern Ohio Alcohol Center, Department of Inflammation and Immunity, Cleveland Clinic, Cleveland, OH, United States.
Department of Gastroenterology and Hepatology, Cleveland Clinic, Cleveland, OH, United States.
Front Endocrinol (Lausanne). 2023 Dec 15;14:1267996. doi: 10.3389/fendo.2023.1267996. eCollection 2023.
The RIP1-RIP3-MLKL-mediated cell death pathway is associated with progression of non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Previous work identified a critical role for MLKL, the key effector regulating necroptosis, but not RIP3, in mediating high fat diet-induced liver injury in mice. RIP1 and RIP3 have active N-terminus kinase domains essential for activation of MLKL and subsequent necroptosis. However, little is known regarding domain-specific roles of RIP1/RIP3 kinase in liver diseases. Here, we hypothesized that RIP1/RIP3 kinase activity are required for the development of high fat diet-induced liver injury.
and kinase-dead mice on a C57BL/6J background and their littermate controls (WT) were allowed free access to a diet high in fat, fructose and cholesterol (FFC diet) or chow diet.
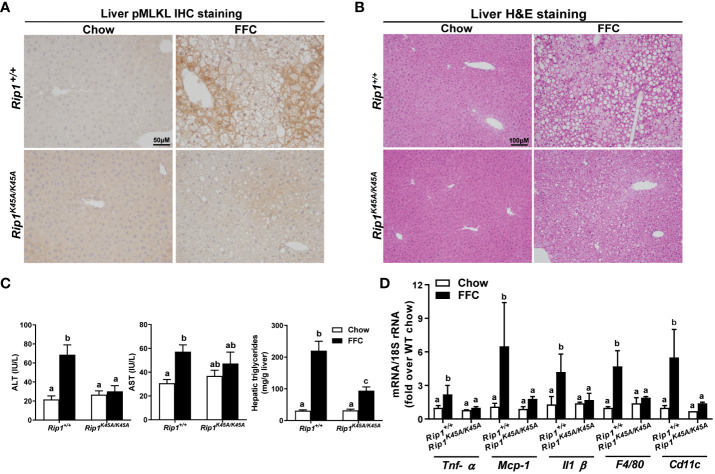
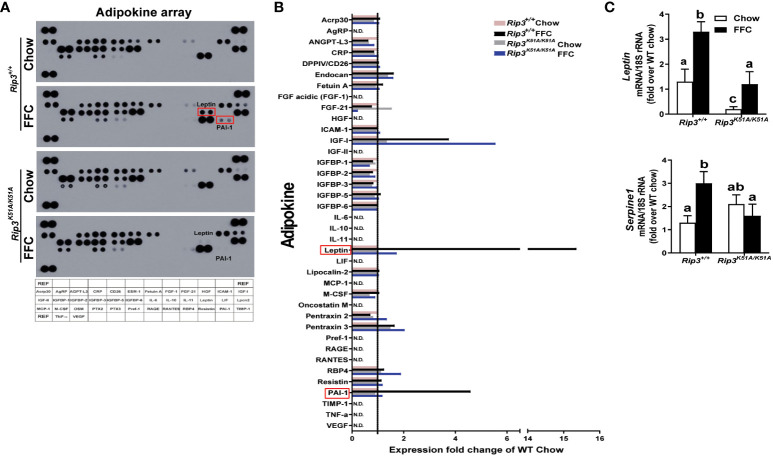
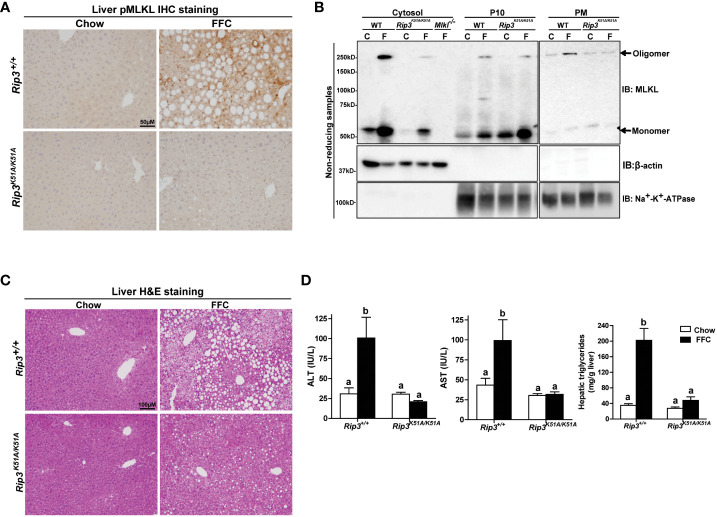
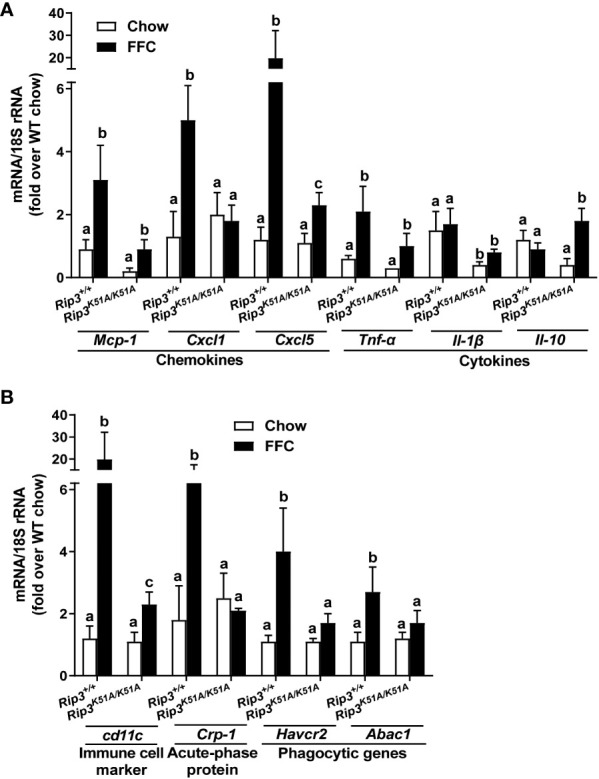
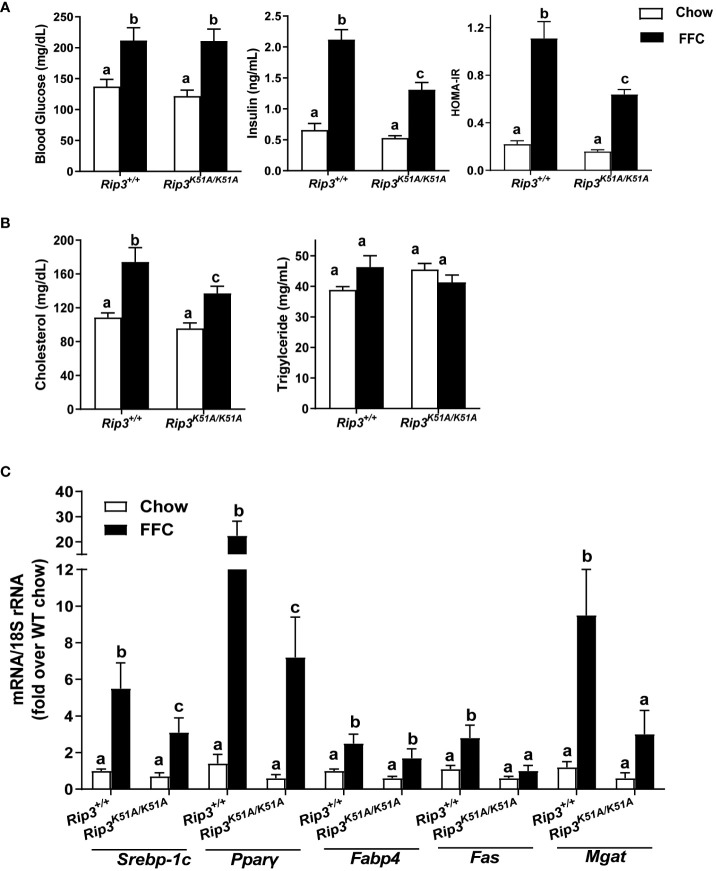
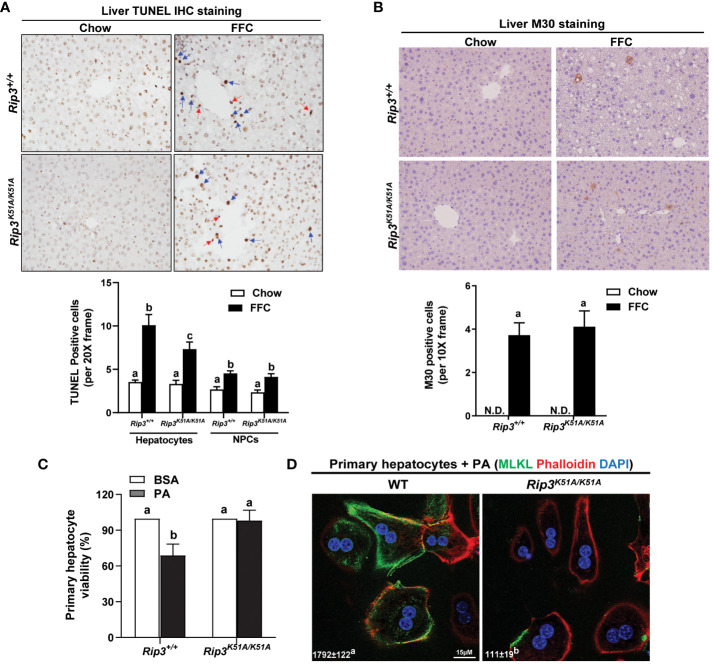
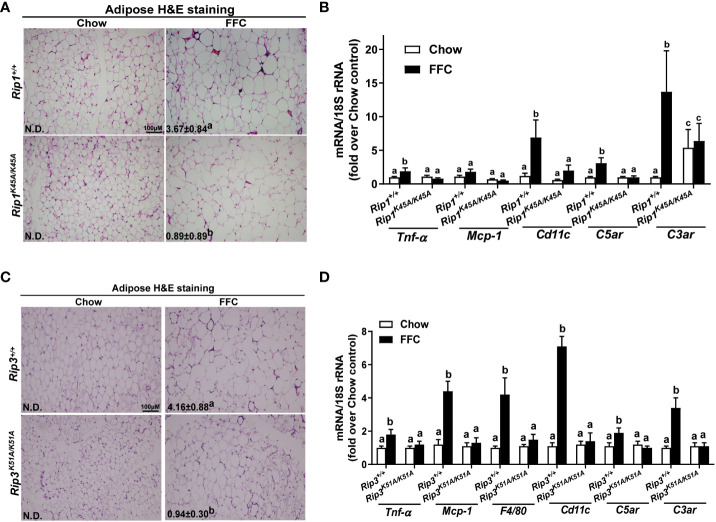
Both and mice were protected against FFC diet-induced steatosis, hepatocyte injury and expression of hepatic inflammatory cytokines and chemokines. FFC diet increased phosphorylation and oligomerization of MLKL and hepatocyte death in livers of WT, but not in , mice. Consistent with data, RIP3 kinase deficiency in primary hepatocytes prevented palmitic acid-induced translocation of MLKL to the cell surface and cytotoxicity. Additionally, loss of or kinase suppressed FFC diet-mediated formation of crown-like structures (indicators of dead adipocytes) and expression of mRNA for inflammatory response genes in epididymal adipose tissue. Moreover, FFC diet increased expression of multiple adipokines, including leptin and plasminogen activator inhibitor 1, in WT mice, which was abrogated by kinase deficiency.
The current data indicate that both RIP1 and RIP3 kinase activity contribute to FFC diet-induced liver injury. This effect of RIP1 and RIP3 kinase deficiency on injury is consistent with the protection of mice from high fat diet-induced liver injury, but not the reported lack of protection in mice. Taken together with previous reports, our data suggest that other domains of RIP3 likely counteract the effect of RIP3 kinase in response to high fat diets.
RIP1-RIP3-MLKL 介导的细胞死亡途径与非酒精性脂肪性肝炎/脂肪性肝炎(NAFL/NASH)的进展有关。先前的工作确定了 MLKL 的关键作用,MLKL 是调节坏死性凋亡的关键效应因子,但 RIP3 不是,它在介导高脂肪饮食诱导的小鼠肝损伤中起作用。RIP1 和 RIP3 具有活性 N 端激酶结构域,对于 MLKL 的激活和随后的坏死性凋亡至关重要。然而,关于 RIP1/RIP3 激酶在肝脏疾病中的特定结构域作用知之甚少。在这里,我们假设 RIP1/RIP3 激酶活性是高脂肪饮食诱导的肝损伤发展所必需的。
和激酶缺陷型小鼠在 C57BL/6J 背景下及其同窝对照(WT)允许自由访问高脂肪、果糖和胆固醇饮食(FFC 饮食)或标准饮食。
和小鼠均能抵抗 FFC 饮食诱导的脂肪变性、肝细胞损伤以及肝炎性细胞因子和趋化因子的表达。FFC 饮食增加了 WT 肝脏中 MLKL 的磷酸化和寡聚化以及肝细胞死亡,但在小鼠中则不然。与数据一致,在原代肝细胞中 RIP3 激酶缺陷阻止了 MLKL 向细胞表面的易位和细胞毒性。此外,缺失或激酶抑制了 FFC 饮食介导的冠状结构(死脂肪细胞的指标)形成和附睾脂肪组织中炎症反应基因的表达。此外,FFC 饮食增加了 WT 小鼠中多种脂肪因子的表达,包括瘦素和纤溶酶原激活物抑制剂 1,而这种表达被激酶缺陷所消除。
目前的数据表明,RIP1 和 RIP3 激酶活性都有助于 FFC 饮食诱导的肝损伤。RIP1 和 RIP3 激酶缺失对损伤的影响与小鼠对高脂肪饮食诱导的肝损伤的保护作用一致,但与报告中的小鼠缺乏保护作用不一致。结合以前的报道,我们的数据表明,RIP3 的其他结构域可能在应对高脂肪饮食时抵消了 RIP3 激酶的作用。