Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences, Tehran, Iran.
Cardiogenetic Research Center, Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences, Tehran, Iran.
BMC Cardiovasc Disord. 2024 Jan 2;24(1):1. doi: 10.1186/s12872-023-03676-z.
Primary carnitine deficiency (PCD) denotes low carnitine levels with an autosomal recessive pattern of inheritance. Cardiomyopathy is the most common cardiac symptom in patients with PCD, and early diagnosis can prevent complications. Next-generation sequencing can identify genetic variants attributable to PCD efficiently.
We aimed to detect the genetic cause of the early manifestations of hypertrophic cardiomyopathy and metabolic abnormalities in an Iranian family.
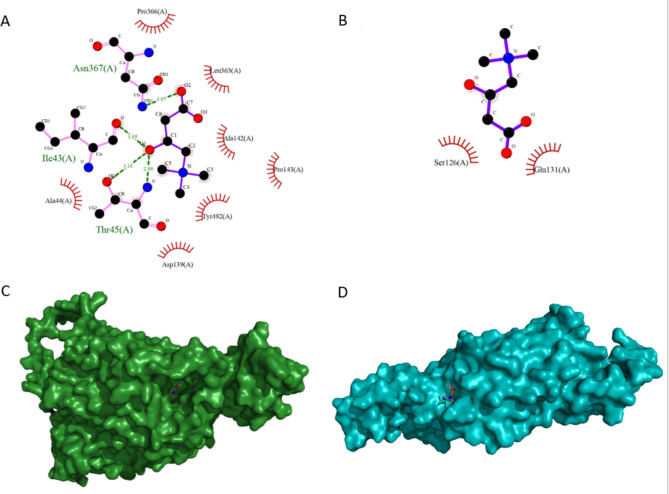
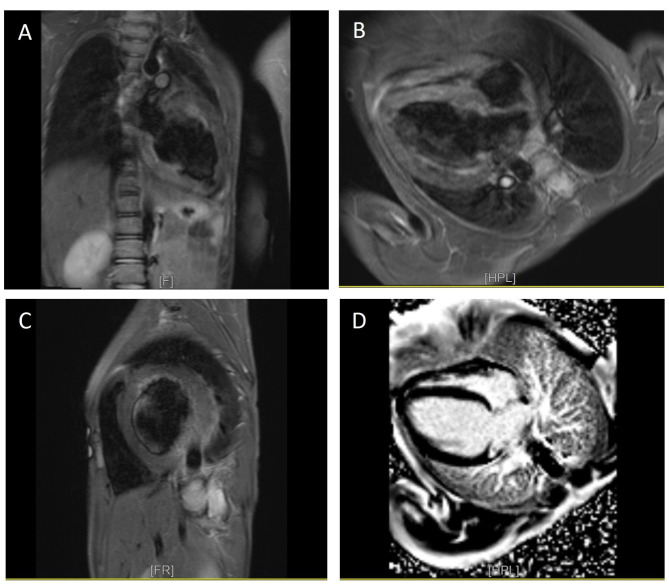
We herein describe an 8-year-old boy with symptoms of weakness and lethargy diagnosed with PCD through clinical evaluations, lab tests, echocardiography, and cardiac magnetic resonance imaging. The candidate variant was confirmed through whole-exome sequencing, polymerase chain reaction, and direct Sanger sequencing. The binding efficacy of normal and mutant protein-ligand complexes were evaluated via structural modeling and docking studies.
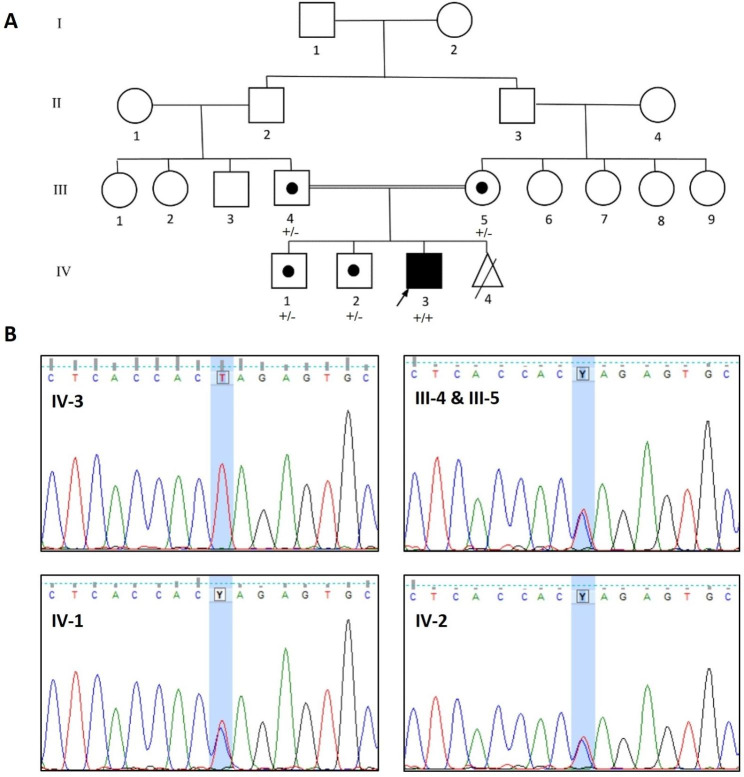
Clinical evaluations, echocardiography, and cardiac magnetic resonance imaging findings revealed hypertrophic cardiomyopathy as a clinical presentation of PCD. Whole-exome sequencing identified a new homozygous variant, SLC22A5 (NM_003060.4), c.821G > A: p.Trp274Ter, associated with carnitine transport. Docking analysis highlighted the impact of the variant on carnitine transport, further indicating its potential role in PCD development.
The c.821G > A: p.Trp274Ter variant in SLC22A5 potentially acted as a pathogenic factor by reducing the binding affinity of organic carnitine transporter type 2 proteins for carnitine. So, the c.821G > A variant may be associated with carnitine deficiency, metabolic abnormalities, and cardiomyopathic characteristics.
原发性肉碱缺乏症(PCD)是一种常染色体隐性遗传的肉碱水平降低的疾病。心肌病是 PCD 患者最常见的心脏症状,早期诊断可以预防并发症。下一代测序可以有效地识别导致 PCD 的遗传变异。
我们旨在检测一个伊朗家庭中肥厚型心肌病和代谢异常的早期表现的遗传原因。
我们在此描述了一名 8 岁男孩,他因乏力和嗜睡等症状被诊断为 PCD,通过临床评估、实验室检查、超声心动图和心脏磁共振成像。候选变体通过全外显子组测序、聚合酶链反应和直接 Sanger 测序得到确认。通过结构建模和对接研究评估正常和突变蛋白-配体复合物的结合效力。
临床评估、超声心动图和心脏磁共振成像结果显示肥厚型心肌病是 PCD 的临床表现。全外显子组测序发现了一个新的纯合变异,SLC22A5(NM_003060.4),c.821G > A:p.Trp274Ter,与肉碱转运有关。对接分析突出了该变异对肉碱转运的影响,进一步表明其在 PCD 发展中的潜在作用。
SLC22A5 中的 c.821G > A:p.Trp274Ter 变异可能通过降低有机肉碱转运体 2 蛋白对肉碱的结合亲和力,作为一个致病因素。因此,c.821G > A 变异可能与肉碱缺乏、代谢异常和心肌病特征有关。