Department of Clinical Genetics, Erasmus MC University Medical Center, Rotterdam, The Netherlands.
Institute of Human Genetics, Universitätsklinikum Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91054, Erlangen, Germany.
Eur J Hum Genet. 2024 Mar;32(3):350-356. doi: 10.1038/s41431-023-01530-6. Epub 2024 Jan 10.
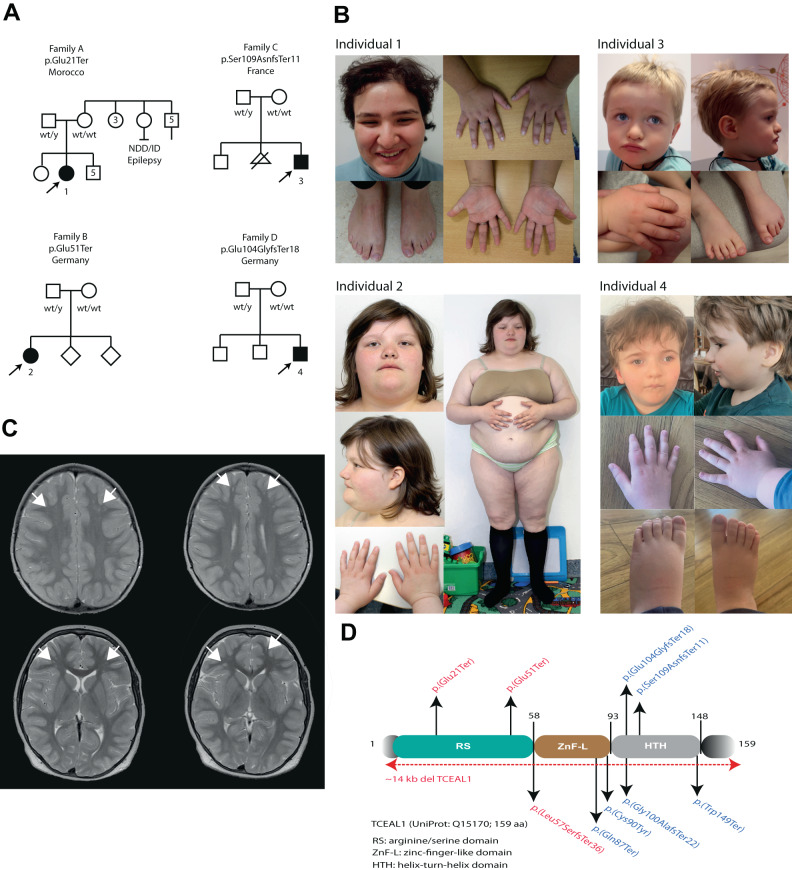
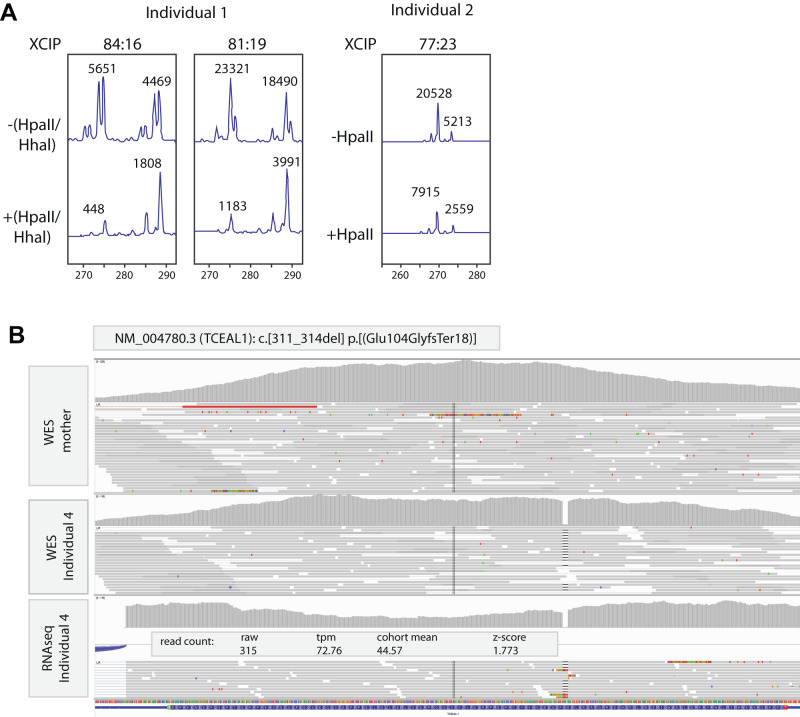
Numerous contiguous gene deletion syndromes causing neurodevelopmental disorders have previously been defined using cytogenetics for which only in the current genomic era the disease-causing genes have become elucidated. One such example is deletion at Xq22.2, previously associated with a neurodevelopmental disorder which has more recently been found to be caused by de novo loss-of-function variants in TCEAL1. So far, a single study reported six unrelated individuals with this monogenetic disorder, presenting with syndromic features including developmental delay especially affecting expressive speech, intellectual disability, autistic-like behaviors, hypotonia, gait abnormalities and mild facial dysmorphism, in addition to ocular, gastrointestinal, and immunologic abnormalities. Here we report on four previously undescribed individuals, including two adults, with de novo truncating variants in TCEAL1, identified through trio exome or genome sequencing, further delineating the phenotype of the TCEAL1-related disorder. Whereas overall we identify similar features compared to the original report, we also highlight features in our adult individuals including hyperphagia, obesity, and endocrine abnormalities including hyperinsulinemia, hyperandrogenemia, and polycystic ovarian syndrome. X chromosome inactivation and RNA-seq studies further provide functional insights in the molecular mechanisms. Together this report expands the phenotypic and molecular spectrum of the TCEAL1-related disorder which will be useful for counseling of newly identified individuals and their families.
许多导致神经发育障碍的连续基因缺失综合征以前都是通过细胞遗传学来定义的,而只有在当前的基因组时代,这些疾病的致病基因才被阐明。Xq22.2 缺失就是一个例子,它以前与一种神经发育障碍有关,而最近发现这种障碍是由 TCEAL1 中的新生失活功能变异引起的。到目前为止,只有一项研究报道了 6 名患有这种单基因疾病的无血缘关系的个体,他们表现出综合征特征,包括发育迟缓,特别是影响表达性语言、智力残疾、类自闭症行为、张力减退、步态异常和轻度面部畸形,此外还有眼部、胃肠道和免疫异常。在这里,我们报告了 4 名以前未描述的个体,包括 2 名成年人,他们携带 TCEAL1 的新生截断变异,这些变异是通过三体外显子或基因组测序发现的,进一步描述了 TCEAL1 相关疾病的表型。虽然我们总体上与原始报告相比发现了类似的特征,但我们也强调了我们的成年个体中的特征,包括食欲过盛、肥胖和内分泌异常,包括高胰岛素血症、高雄激素血症和多囊卵巢综合征。X 染色体失活和 RNA-seq 研究进一步提供了分子机制的功能见解。本报告扩展了 TCEAL1 相关疾病的表型和分子谱,这将有助于对新发现的个体及其家属进行咨询。