Danilov Sergei M, Adzhubei Ivan A, Kozuch Alexander J, Petukhov Pavel A, Popova Isolda A, Choudhury Ananyo, Sengupta Dhriti, Dudek Steven M
Department of Medicine, Division of Pulmonary, Critical Care, Sleep and Allergy, University of Illinois Chicago, Chicago, IL 60612, USA.
Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA.
Biomedicines. 2024 Jan 12;12(1):162. doi: 10.3390/biomedicines12010162.
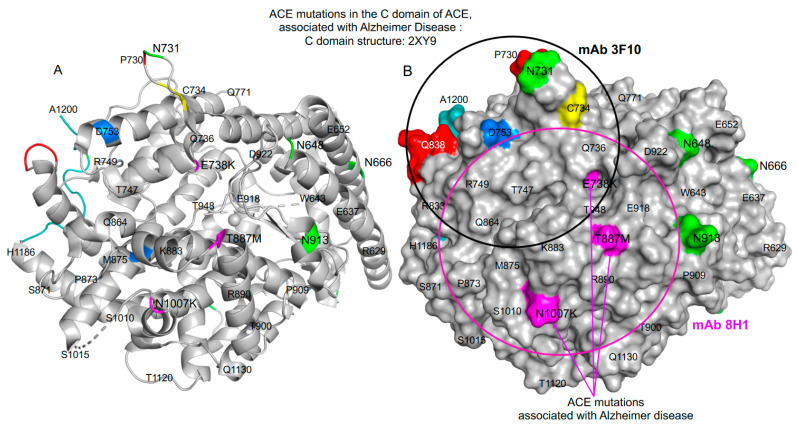
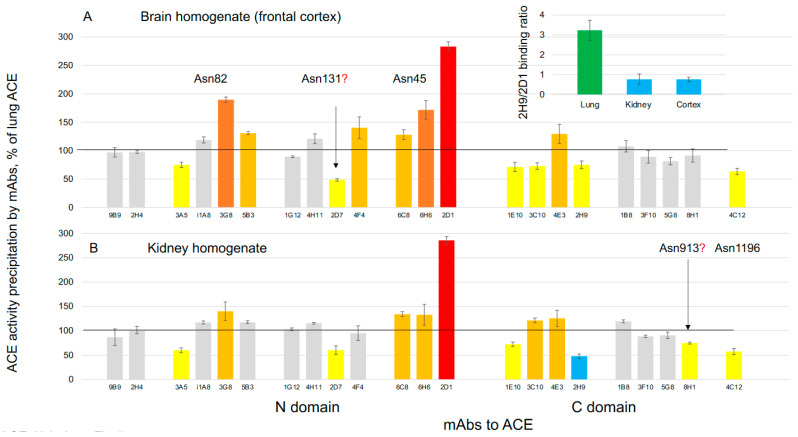
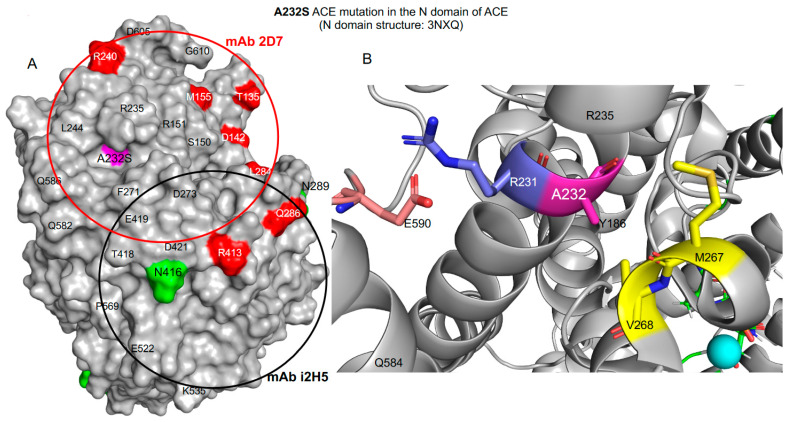
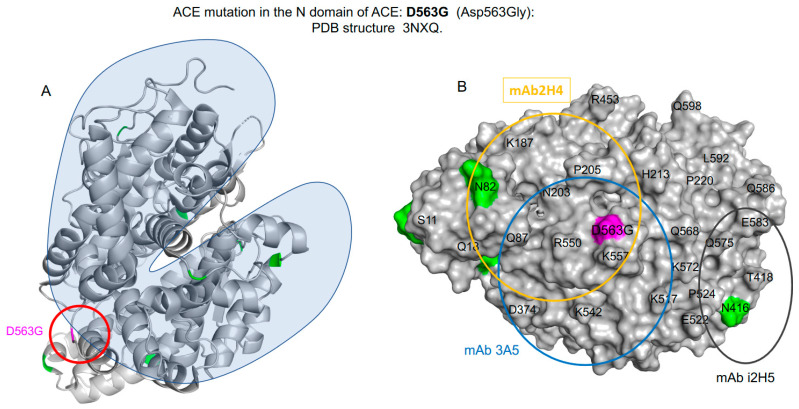
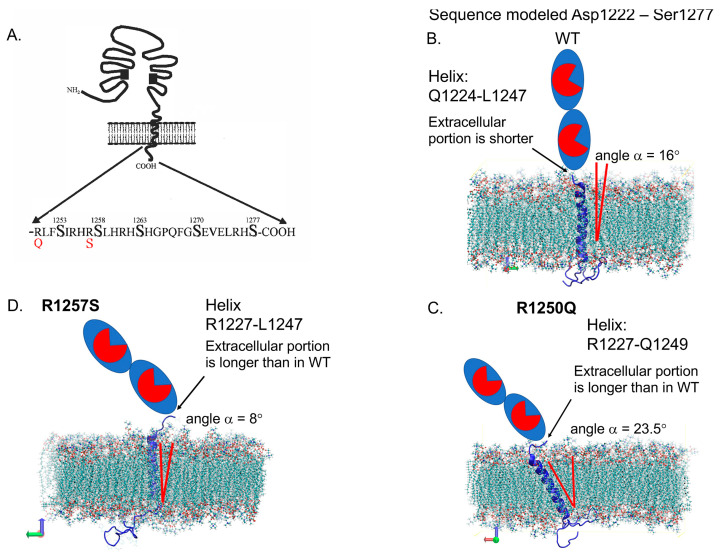
We hypothesized that subjects with heterozygous loss-of-function (LoF) mutations are at risk for Alzheimer's disease because amyloid Aβ42, a primary component of the protein aggregates that accumulate in the brains of AD patients, is cleaved by ACE (angiotensin I-converting enzyme). Thus, decreased ACE activity in the brain, either due to genetic mutation or the effects of ACE inhibitors, could be a risk factor for AD. To explore this hypothesis in the current study, existing SNP databases were analyzed for LoF mutations using four predicting tools, including PolyPhen-2, and compared with the topology of known mutations already associated with AD. The combined frequency of >400 of these LoF-damaging mutations in the general population is quite significant-up to 5%-comparable to the frequency of AD in the population > 70 y.o., which indicates that the contribution of low ACE in the development of AD could be under appreciated. Our analysis suggests several mechanisms by which ACE mutations may be associated with Alzheimer's disease. Systematic analysis of blood ACE levels in patients with all mutations is likely to have clinical significance because available sequencing data will help detect persons with increased risk of late-onset Alzheimer's disease. Patients with transport-deficient mutations (about 20% of damaging ACE mutations) may benefit from preventive or therapeutic treatment with a combination of chemical and pharmacological (e.g., centrally acting ACE inhibitors) chaperones and proteosome inhibitors to restore impaired surface ACE expression, as was shown previously by our group for another transport-deficient ACE mutation-Q1069R.
我们推测,具有杂合功能丧失(LoF)突变的个体有患阿尔茨海默病的风险,因为淀粉样β-蛋白42(Aβ42)是在阿尔茨海默病患者大脑中积累的蛋白质聚集体的主要成分,可被血管紧张素转换酶(ACE)切割。因此,由于基因突变或ACE抑制剂的作用导致大脑中ACE活性降低,可能是阿尔茨海默病的一个风险因素。为了在当前研究中探索这一假设,我们使用包括PolyPhen-2在内的四种预测工具,对现有的单核苷酸多态性(SNP)数据库进行了LoF突变分析,并与已经与阿尔茨海默病相关的已知突变拓扑结构进行了比较。在普通人群中,这些具有LoF损伤性的400多个突变的综合频率相当高——高达5%——与70岁以上人群中阿尔茨海默病的发病率相当,这表明低ACE水平在阿尔茨海默病发展中的作用可能未得到充分认识。我们的分析表明了几种ACE突变可能与阿尔茨海默病相关的机制。对所有突变患者的血液ACE水平进行系统分析可能具有临床意义,因为现有的测序数据将有助于检测出晚发性阿尔茨海默病风险增加的人群。具有转运缺陷突变的患者(约占ACE损伤性突变的20%)可能受益于化学和药理学伴侣(如中枢作用的ACE抑制剂)和蛋白酶体抑制剂联合的预防性或治疗性治疗,以恢复受损的表面ACE表达,正如我们小组之前针对另一种转运缺陷型ACE突变——Q1069R所显示的那样。