Avloniti Maria, Evangelidou Maria, Gomini Maria, Loupis Theodore, Emmanouil Mary, Mitropoulou Adamantia, Tselios Theodore, Lassmann Hans, Gruart Agnès, Delgado-García José M, Probert Lesley, Kyrargyri Vasiliki
Laboratory of Molecular Genetics, Hellenic Pasteur Institute, Athens, Greece.
Greek Genome Centre, Biomedical Research Foundation of the Academy of Athens (BRFAA), Athens, Greece.
J Neuroinflammation. 2024 Jan 27;21(1):34. doi: 10.1186/s12974-024-03023-9.
Multiple sclerosis (MS) is a neuroinflammatory demyelinating disease characterized by motor deficits and cognitive decline. Many immune aspects of the disease are understood through studies in the experimental autoimmune encephalomyelitis (EAE) model, including the contribution of the NF-κB transcription factor to neuroinflammation. However, the cell-specific roles of NF-κB to EAE and its cognitive comorbidities still needs further investigation. We have previously shown that the myeloid cell NF-κB plays a role in the healthy brain by exerting homeostatic regulation of neuronal excitability and synaptic plasticity and here we investigated its role in EAE.
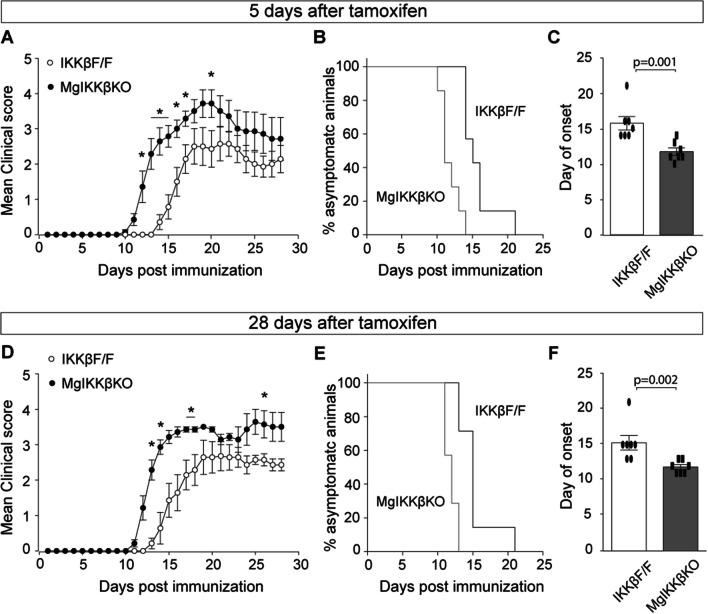
We used constitutive MφIKKβΚΟ mice, in which depletion of IKKβ, the main activating kinase of NF-κB, was global to CNS and peripheral macrophages, and ΜgΙΚΚβKO mice, in which depletion was inducible and specific to CNS macrophages by 28 days after tamoxifen administration. We subjected these mice to MOG induced EAE and cuprizone-induced demyelination. We measured pathology by immunohistochemistry, investigated molecular mechanisms by RNA sequencing analysis and studied neuronal functions by in vivo electrophysiology in awake animals.
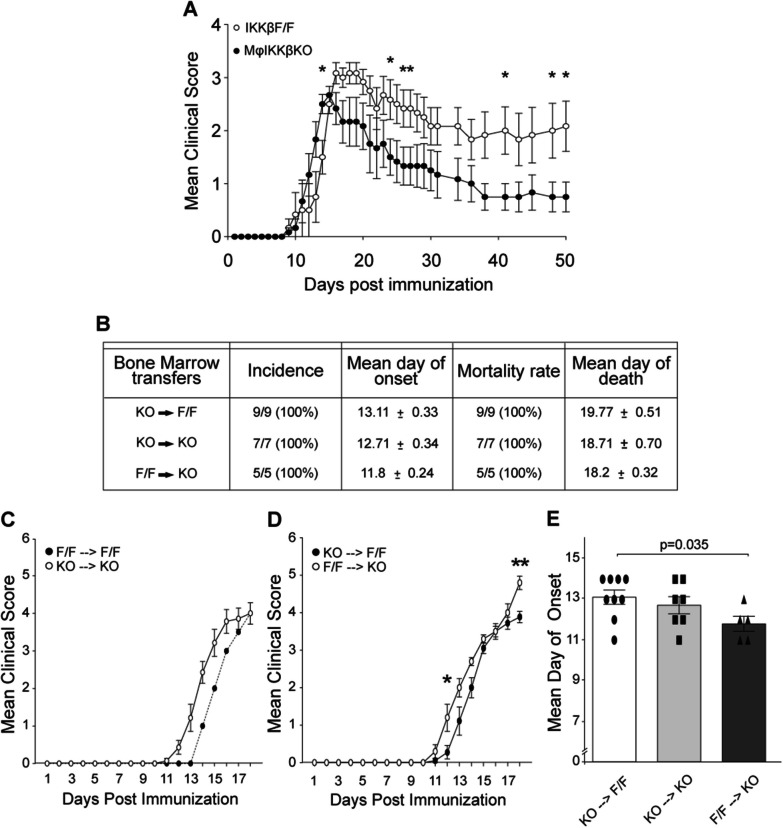
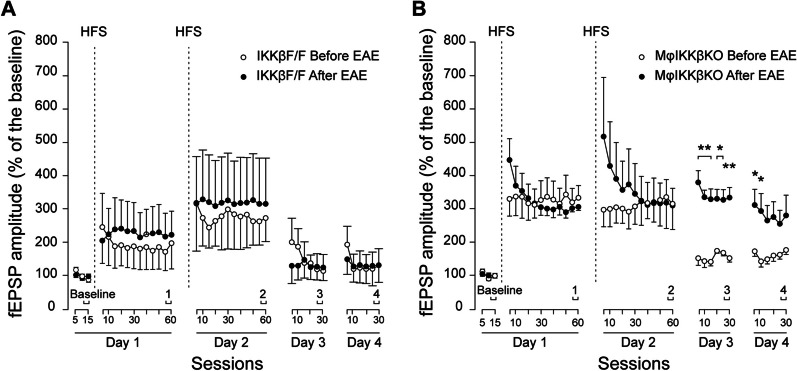
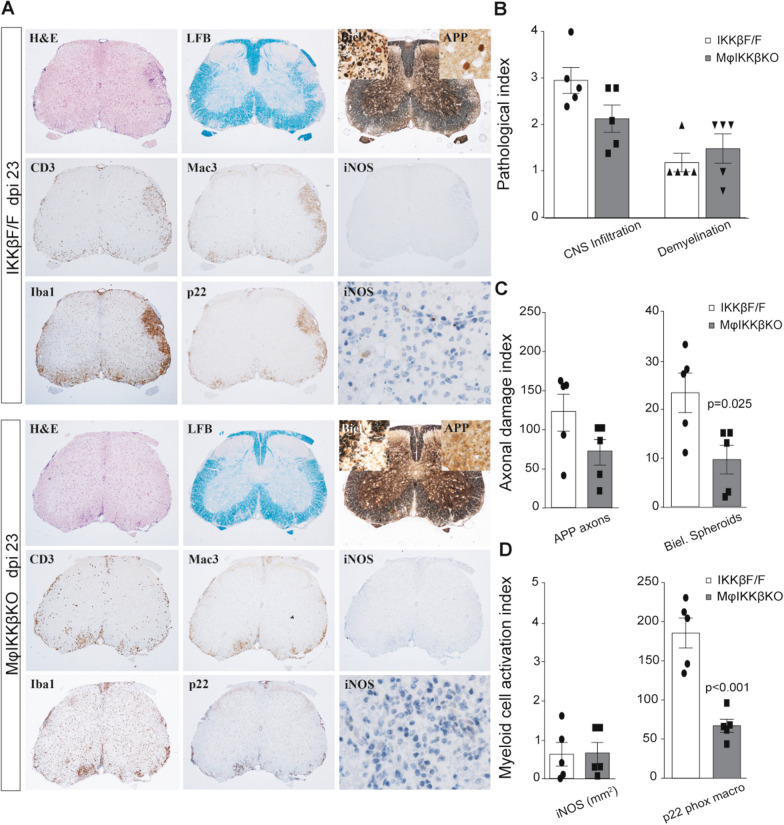
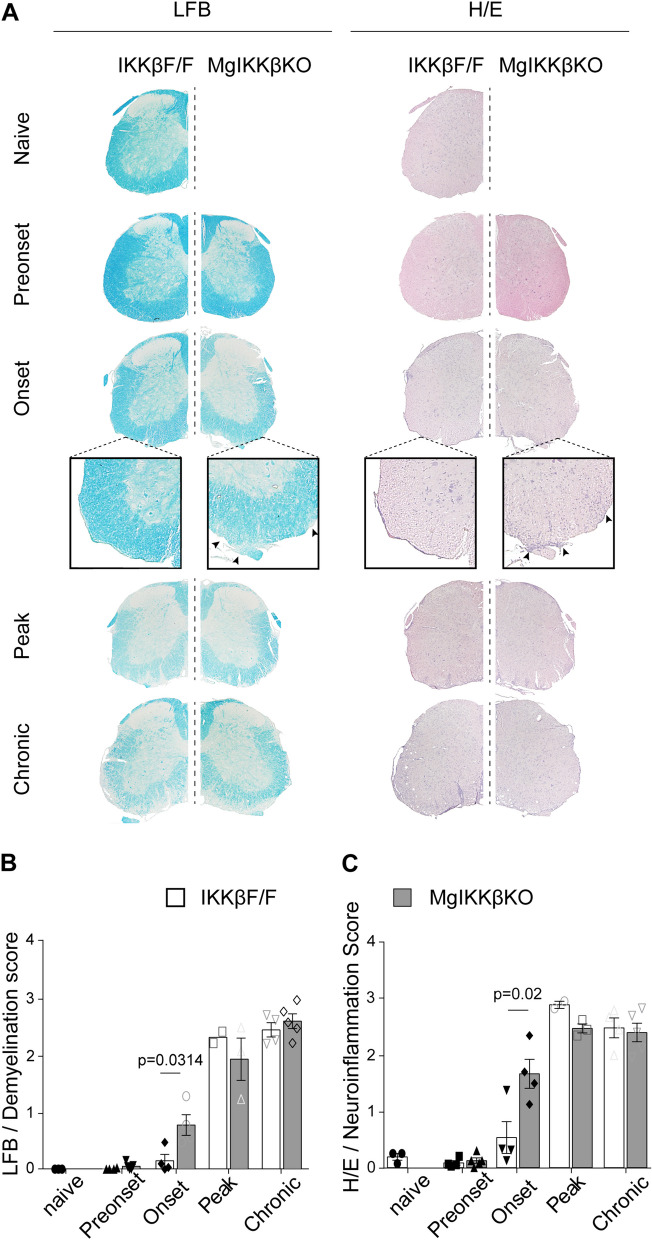
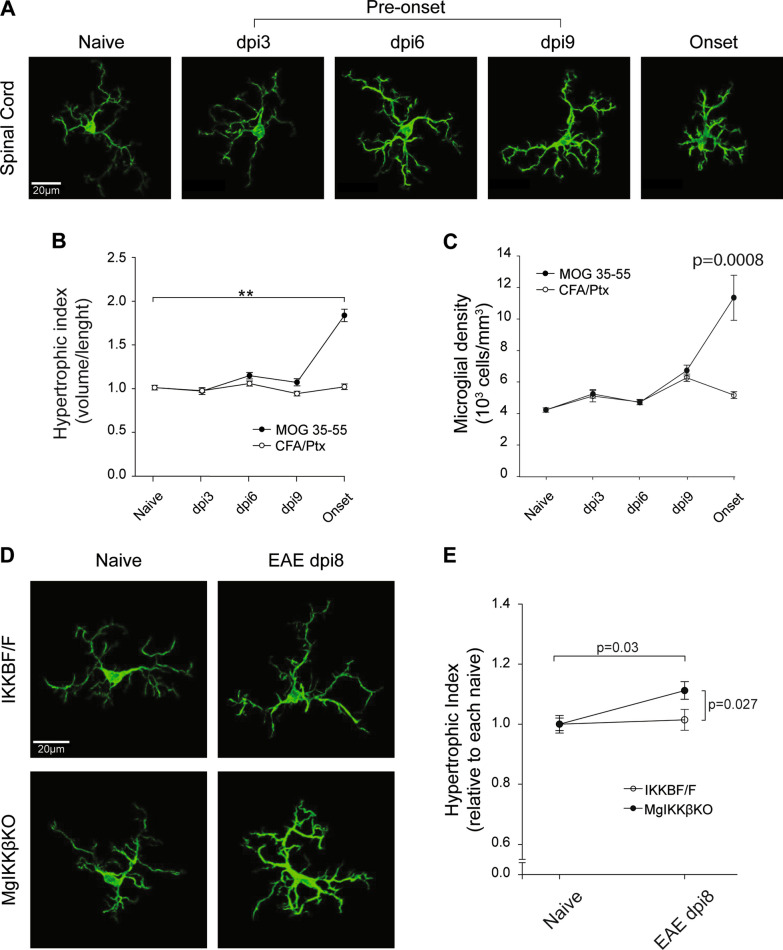
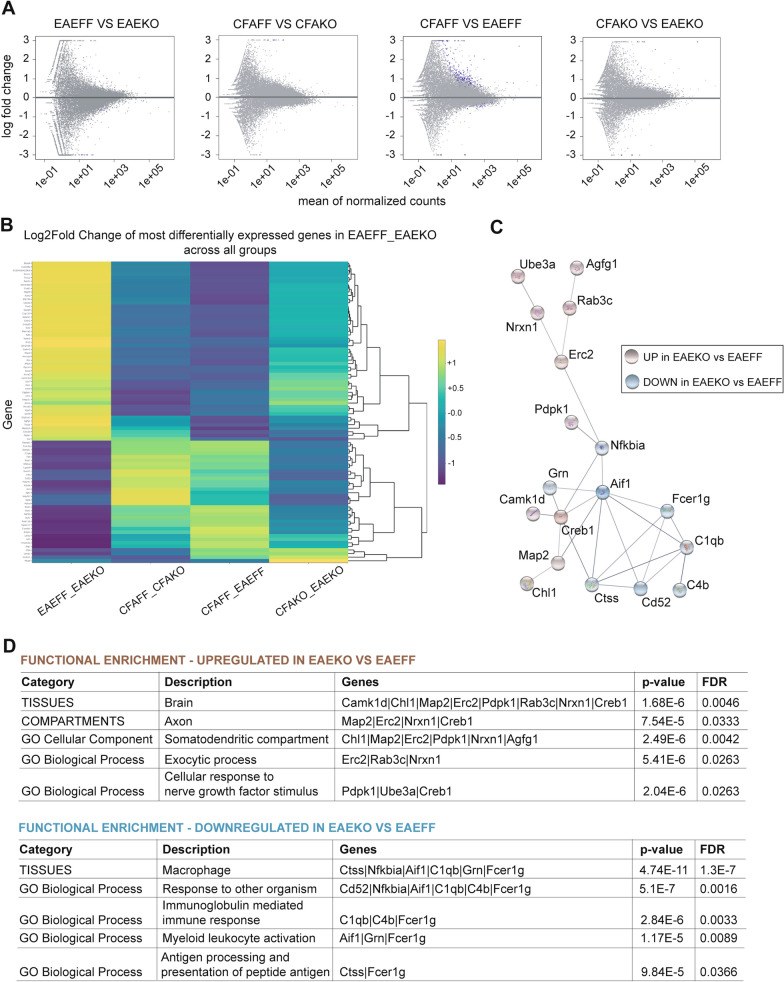
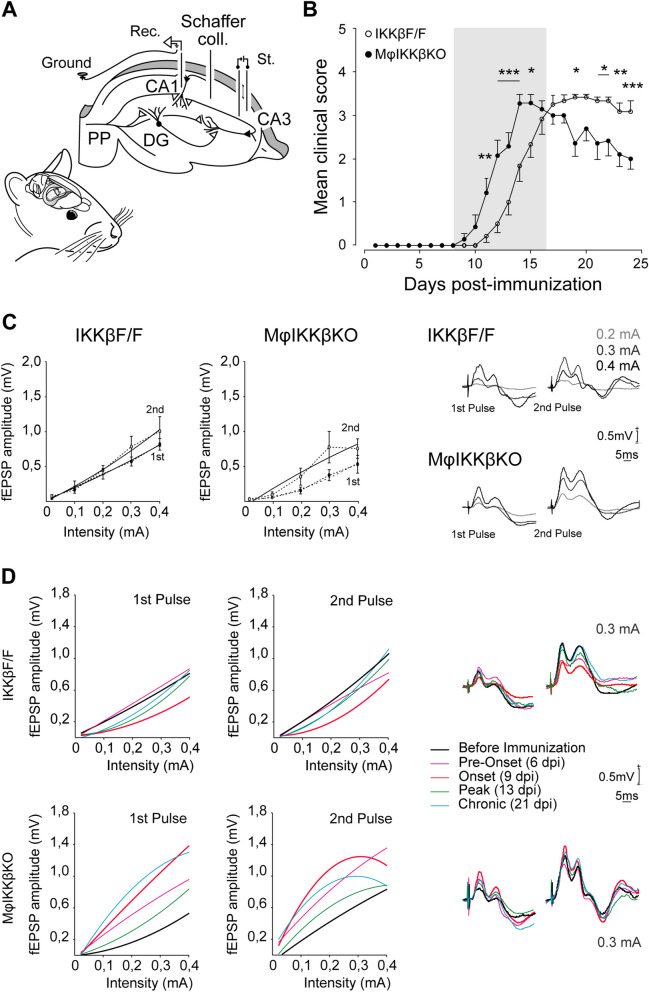
Global depletion of IKKβ from myeloid cells in MφIKKβΚΟ mice accelerated the onset and significantly supressed chronic EAE. Knocking out IKKβ only from CNS resident macrophages accelerated the onset and exacerbated chronic EAE, accompanied by earlier demyelination and immune cell infiltration but had no effect in cuprizone-induced demyelination. Peripheral T cell effector functions were not affected by myeloid cell deletion of IKKβ, but CNS resident mechanisms, such as microglial activation and neuronal hyperexcitability were altered from early in EAE. Lastly, depletion of myeloid cell IKKβ resulted in enhanced late long-term potentiation in EAE.
IKKβ-mediated activation of NF-κΒ in myeloid cells has opposing roles in EAE depending on the cell type and the disease stage. In CNS macrophages it is protective while in peripheral macrophages it is disease-promoting and acts mainly during chronic disease. Although clinically protective, CNS myeloid cell IKKβ deletion dysregulates neuronal excitability and synaptic plasticity in EAE. These effects of IKKβ on brain cognitive abilities deserve special consideration when therapeutic interventions that inhibit NF-κB are used in MS.
多发性硬化症(MS)是一种神经炎性脱髓鞘疾病,其特征为运动功能障碍和认知功能衰退。通过对实验性自身免疫性脑脊髓炎(EAE)模型的研究,人们对该疾病的许多免疫方面有了一定了解,包括核因子κB(NF-κB)转录因子在神经炎症中的作用。然而,NF-κB在EAE及其认知合并症中的细胞特异性作用仍需进一步研究。我们之前已经表明,髓样细胞NF-κB通过对神经元兴奋性和突触可塑性进行稳态调节,在健康大脑中发挥作用,在此我们研究了其在EAE中的作用。
我们使用了组成型MφIKKβΚΟ小鼠,其中NF-κB的主要激活激酶IKKβ在中枢神经系统(CNS)和外周巨噬细胞中均被整体敲除;以及ΜgΙΚΚβKO小鼠,其中IKKβ的敲除在给予他莫昔芬后28天可诱导且特异性针对CNS巨噬细胞。我们使这些小鼠发生髓鞘少突胶质细胞糖蛋白(MOG)诱导的EAE和铜离子螯合剂诱导的脱髓鞘。我们通过免疫组织化学测量病理学变化,通过RNA测序分析研究分子机制,并通过对清醒动物进行体内电生理学研究神经元功能。
MφIKKβΚΟ小鼠中髓样细胞IKKβ的整体缺失加速了EAE的发病,并显著抑制了慢性EAE。仅从CNS驻留巨噬细胞中敲除IKKβ加速了发病并加重了慢性EAE,伴有更早的脱髓鞘和免疫细胞浸润,但对铜离子螯合剂诱导的脱髓鞘没有影响。外周T细胞效应功能不受髓样细胞IKKβ缺失的影响,但CNS驻留机制,如小胶质细胞激活和神经元兴奋性过高,在EAE早期就发生了改变。最后,髓样细胞IKKβ的缺失导致EAE中晚期长时程增强作用增强。
IKKβ介导的髓样细胞中NF-κΒ的激活在EAE中根据细胞类型和疾病阶段具有相反的作用。在CNS巨噬细胞中它具有保护作用,而在外周巨噬细胞中它促进疾病发展,且主要在慢性疾病期间起作用。虽然在临床上具有保护作用,但CNS髓样细胞IKKβ的缺失会在EAE中失调神经元兴奋性和突触可塑性。当在MS中使用抑制NF-κB的治疗干预措施时,IKKβ对大脑认知能力的这些影响值得特别考虑。