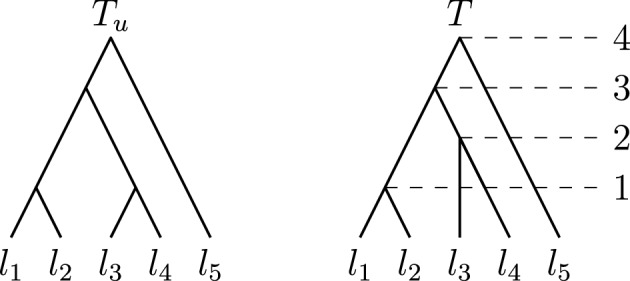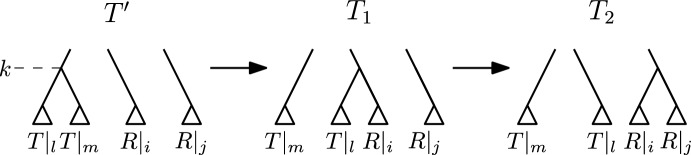Biological Data Science Laboratory, School of Mathematics and Statistics, University of Canterbury, Christchurch, New Zealand.
Faculty of Computer Science, Dalhousie University, Halifax, Canada.
Bull Math Biol. 2024 Jan 31;86(3):24. doi: 10.1007/s11538-023-01244-2.
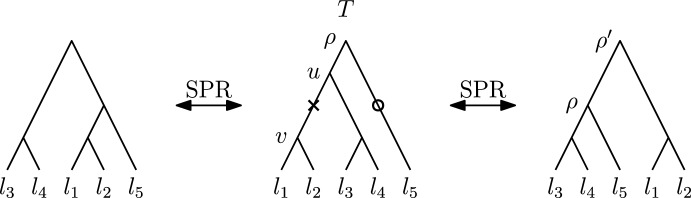
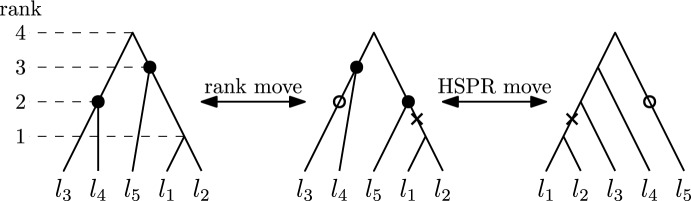
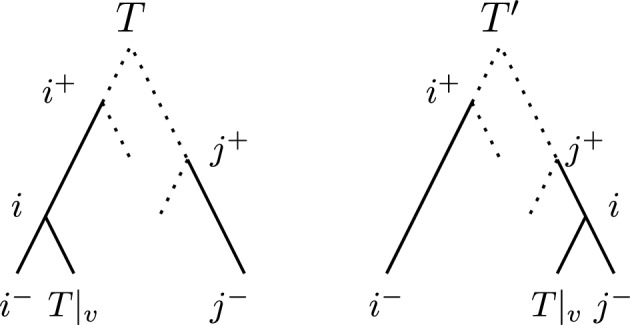
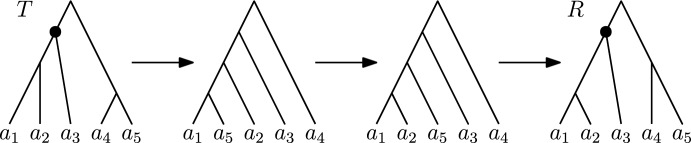
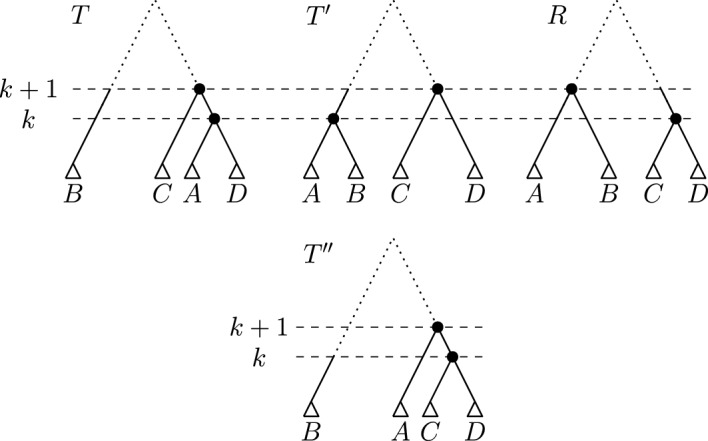
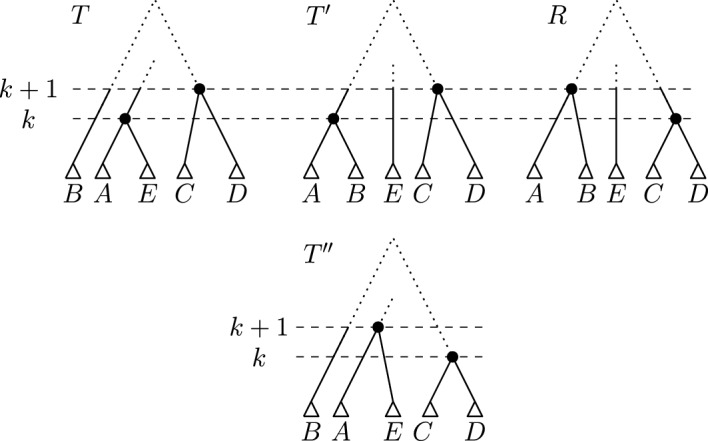
Phylogenetic trees are a mathematical formalisation of evolutionary histories between organisms, species, genes, cancer cells, etc. For many applications, e.g. when analysing virus transmission trees or cancer evolution, (phylogenetic) time trees are of interest, where branch lengths represent times. Computational methods for reconstructing time trees from (typically molecular) sequence data, for example Bayesian phylogenetic inference using Markov Chain Monte Carlo (MCMC) methods, rely on algorithms that sample the treespace. They employ tree rearrangement operations such as [Formula: see text] (Subtree Prune and Regraft) and [Formula: see text] (Nearest Neighbour Interchange) or, in the case of time tree inference, versions of these that take times of internal nodes into account. While the classic [Formula: see text] tree rearrangement is well-studied, its variants for time trees are less understood, limiting comparative analysis for time tree methods. In this paper we consider a modification of the classical [Formula: see text] rearrangement on the space of ranked phylogenetic trees, which are trees equipped with a ranking of all internal nodes. This modification results in two novel treespaces, which we propose to study. We begin this study by discussing algorithmic properties of these treespaces, focusing on those relating to the complexity of computing distances under the ranked [Formula: see text] operations as well as similarities and differences to known tree rearrangement based treespaces. Surprisingly, we show the counterintuitive result that adding leaves to trees can actually decrease their ranked [Formula: see text] distance, which may have an impact on the results of time tree sampling algorithms given uncertain "rogue taxa".
系统发育树是生物、物种、基因、癌细胞等之间进化历史的数学形式化。对于许多应用,例如分析病毒传播树或癌症进化,(系统发育)时间树是感兴趣的,其中分支长度代表时间。从(通常是分子)序列数据重建时间树的计算方法,例如使用马尔可夫链蒙特卡罗(MCMC)方法的贝叶斯系统发育推断,依赖于对树空间进行采样的算法。它们采用树重排操作,例如 [公式:见文本](子树修剪和重新嫁接)和 [公式:见文本](最近邻交换),或者在时间树推断的情况下,考虑到内部节点时间的这些操作的版本。虽然经典的 [公式:见文本] 树重排得到了很好的研究,但针对时间树的变体理解较少,限制了时间树方法的比较分析。在本文中,我们考虑对分级系统发育树空间上的经典 [公式:见文本] 重排进行修改,即配备所有内部节点排序的树。这种修改导致了两个新的树空间,我们建议研究这两个空间。我们通过讨论这些树空间的算法属性开始这项研究,重点是那些与在分级 [公式:见文本] 操作下计算距离的复杂性以及与基于已知树重排的树空间的相似性和差异有关的属性。令人惊讶的是,我们展示了一个反直觉的结果,即向树添加叶子实际上可以减少它们的分级 [公式:见文本] 距离,这可能会对具有不确定“流氓分类群”的时间树采样算法的结果产生影响。