D'Souza Precilla, Farmer Cristan, Johnston Jean, Han Sangwoo T, Adams David, Hartman Adam L, Zein Wadih, Huryn Laryssa A, Solomon Beth, King Kelly, Jordan Christopher, Myles Jennifer, Nicoli Elena-Raluca, Rothermel Caroline E, Algarin Yoliann Mojica, Huang Reyna, Quimby Rachel, Zainab Mosufa, Bowden Sarah, Crowell Anna, Buckley Ashura, Brewer Carmen, Regier Deborah, Brooks Brian, Baker Eva, Vézina Gilbert, Thurm Audrey, Tifft Cynthia J
Office of the Clinical Director and Medical Genetics Branch, National Human Genome Research Institute, 10 Center Drive, Bethesda MD USA.
Neurodevelopmental and Behavioral Phenotyping Service, National Institute of Mental Health, 10 Center Drive, Bethesda MD USA.
medRxiv. 2024 Jan 4:2024.01.04.24300778. doi: 10.1101/2024.01.04.24300778.
GM1 gangliosidosis (GM1) is an ultra-rare lysosomal storage disease caused by pathogenic variants in galactosidase beta 1 (; NM_000404), primarily characterized by neurodegeneration, often in children. There are no approved treatments for GM1, but clinical trials using gene therapy (NCT03952637, NCT04713475) and small molecule substrate inhibitors (NCT04221451) are ongoing. Understanding the natural history of GM1 is essential for timely diagnosis, facilitating better supportive care, and contextualizing the results of therapeutic trials.
Forty-one individuals with type II GM1 (n=17 late infantile and n=24 juvenile onset) participated in a single-site prospective observational study. Here, we describe the results of extensive multisystem assessment batteries, including clinical labs, neuroimaging, physiological exams, and behavioral assessments.
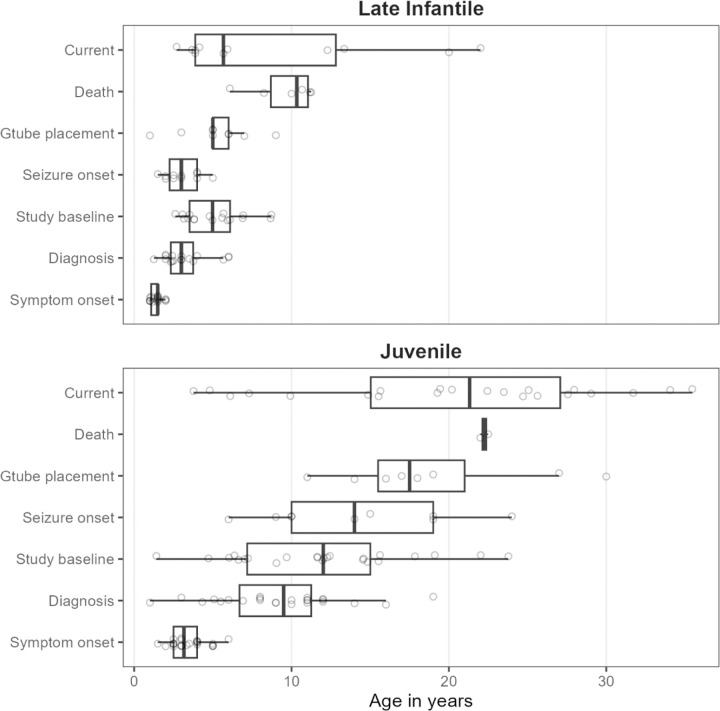
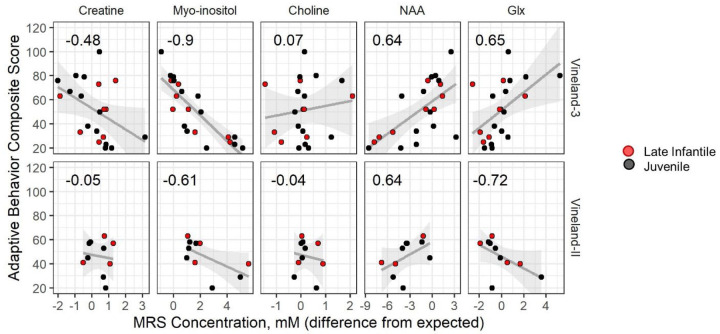
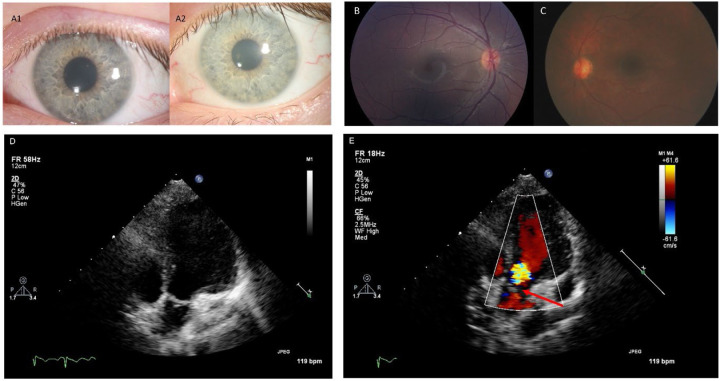
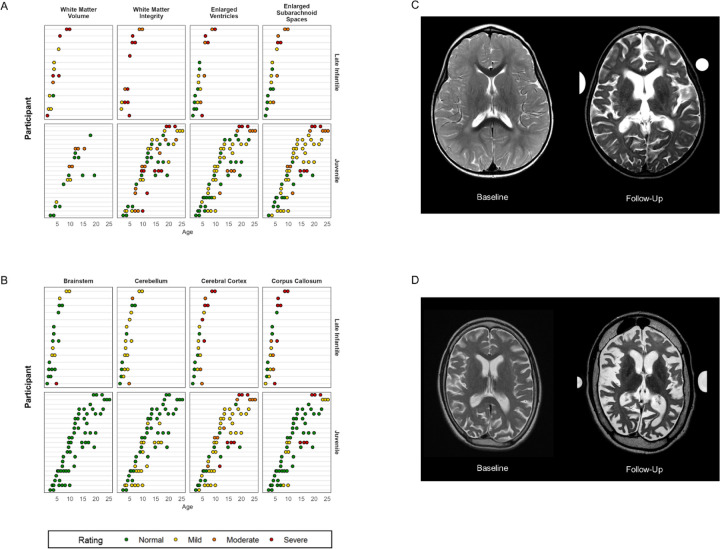
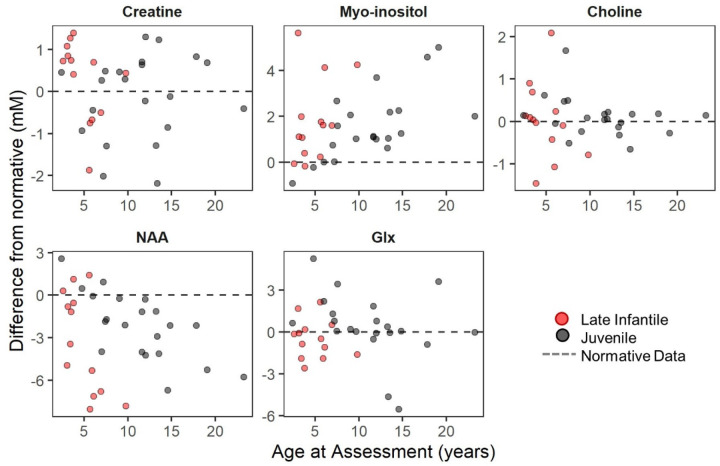
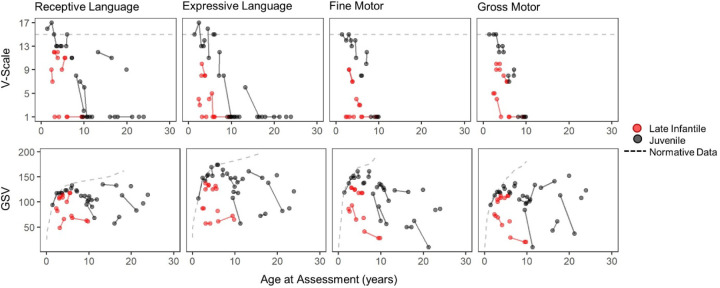
Classification of 37 distinct variants in this cohort was performed according to ACMG criteria and resulted in the upgrade of six and the submission of four new variants to pathogenic or likely pathogenic. In contrast to type I infantile, children with type II disease exhibited normal or near normal hearing and did not have cherry red maculae or significant hepatosplenomegaly. Some older children with juvenile onset developed thickened aortic and/or mitral valves with regurgitation. Serial MRIs demonstrated progressive brain atrophy that were more pronounced in those with late infantile onset. MR spectroscopy showed worsening elevation of myo-inositol and deficit of -acetyl aspartate that were strongly correlated with scores on the Vineland Adaptive Behavior Scale and progress more rapidly in late infantile than juvenile onset disease.
The comprehensive serial phenotyping of type II GM1 patients expands the understanding of disease progression and clarifies some common misconceptions about type II patients. Findings from this 10-year endeavor are a pivotal step toward more timely diagnosis and better supportive care for patients. The wealth of data amassed through this effort will serve as a robust comparator for ongoing and future therapeutic trials.
GM1神经节苷脂贮积症(GM1)是一种极其罕见的溶酶体贮积病,由β-1半乳糖苷酶(; NM_000404)的致病变异引起,主要特征为神经退行性变,常见于儿童。目前尚无获批的GM1治疗方法,但使用基因疗法(NCT03952637、NCT04713475)和小分子底物抑制剂(NCT04221451)的临床试验正在进行。了解GM1的自然病史对于及时诊断、提供更好的支持性护理以及解读治疗试验结果至关重要。
41例II型GM1患者(17例晚婴儿型和24例青少年型)参与了一项单中心前瞻性观察研究。在此,我们描述了广泛的多系统评估结果,包括临床实验室检查、神经影像学检查、生理检查和行为评估。
根据美国医学遗传学与基因组学学会(ACMG)标准对该队列中的37种不同变异进行了分类,导致6种变异升级,并将4种新变异提交为致病或可能致病变异。与I型婴儿型不同,II型疾病患儿的听力正常或接近正常,没有樱桃红斑或明显的肝脾肿大。一些青少年型的大龄儿童出现主动脉瓣和/或二尖瓣增厚并伴有反流。系列磁共振成像(MRI)显示进行性脑萎缩,在晚婴儿型患者中更为明显。磁共振波谱显示肌醇升高和N-乙酰天门冬氨酸缺乏加重,这与文兰适应行为量表得分密切相关,且在晚婴儿型中进展比青少年型更快。
II型GM1患者的全面系列表型分析扩展了对疾病进展的认识,并澄清了一些关于II型患者的常见误解。这项为期10年的研究结果是朝着更及时诊断和为患者提供更好支持性护理迈出的关键一步。通过这项努力积累的大量数据将作为正在进行和未来治疗试验的有力对照。