School of Cancer and Pharmaceutical Sciences, King's College London, 123 Coldharbour Lane, London SE5 9NU, United Kingdom.
Department of Non-Communicable Disease Epidemiology, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, United Kingdom.
Hum Mol Genet. 2024 May 4;33(10):919-929. doi: 10.1093/hmg/ddae014.
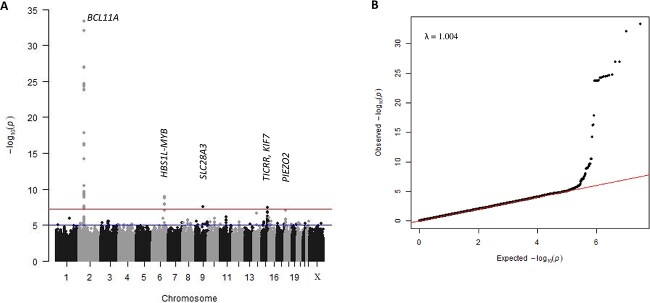
The clinical severity of sickle cell disease (SCD) is strongly influenced by the level of fetal haemoglobin (HbF) persistent in each patient. Three major HbF loci (BCL11A, HBS1L-MYB, and Xmn1-HBG2) have been reported, but a considerable hidden heritability remains. We conducted a genome-wide association study for HbF levels in 1006 Nigerian patients with SCD (HbSS/HbSβ0), followed by a replication and meta-analysis exercise in four independent SCD cohorts (3,582 patients). To dissect association signals at the major loci, we performed stepwise conditional and haplotype association analyses and included public functional annotation datasets. Association signals were detected for BCL11A (lead SNP rs6706648, β = -0.39, P = 4.96 × 10-34) and HBS1L-MYB (lead SNP rs61028892, β = 0.73, P = 1.18 × 10-9), whereas the variant allele for Xmn1-HBG2 was found to be very rare. In addition, we detected three putative new trait-associated regions. Genetically, dissecting the two major loci BCL11A and HBS1L-MYB, we defined trait-increasing haplotypes (P < 0.0001) containing so far unidentified causal variants. At BCL11A, in addition to a haplotype harbouring the putative functional variant rs1427407-'T', we identified a second haplotype, tagged by the rs7565301-'A' allele, where a yet-to-be-discovered causal DNA variant may reside. Similarly, at HBS1L-MYB, one HbF-increasing haplotype contains the likely functional small indel rs66650371, and a second tagged by rs61028892-'C' is likely to harbour a presently unknown functional allele. Together, variants at BCL11A and HBS1L-MYB SNPs explained 24.1% of the trait variance. Our findings provide a path for further investigation of the causes of variable fetal haemoglobin persistence in sickle cell disease.
镰状细胞病(SCD)的临床严重程度受每个患者持续的胎儿血红蛋白(HbF)水平的强烈影响。已经报道了三个主要的 HbF 基因座(BCL11A、HBS1L-MYB 和 Xmn1-HBG2),但仍存在相当大的隐性遗传。我们对 1006 名尼日利亚 SCD(HbSS/HbSβ0)患者的 HbF 水平进行了全基因组关联研究,随后在四个独立的 SCD 队列(3582 名患者)中进行了复制和荟萃分析。为了剖析主要基因座的关联信号,我们进行了逐步条件和单倍型关联分析,并纳入了公共功能注释数据集。在 BCL11A(先导 SNP rs6706648,β=-0.39,P=4.96×10-34)和 HBS1L-MYB(先导 SNP rs61028892,β=0.73,P=1.18×10-9)中检测到关联信号,而 Xmn1-HBG2 的变体等位基因则非常罕见。此外,我们还检测到三个可能的新与性状相关的区域。从遗传学上讲,剖析 BCL11A 和 HBS1L-MYB 这两个主要基因座,我们定义了增加性状的单倍型(P<0.0001),其中包含迄今为止尚未确定的因果变异。在 BCL11A 中,除了包含假定功能变体 rs1427407-T 的单倍型外,我们还鉴定了第二个单倍型,由 rs7565301-A 等位基因标记,可能存在尚未发现的因果 DNA 变异。同样,在 HBS1L-MYB 中,一个增加 HbF 的单倍型包含可能的功能小插入缺失 rs66650371,而由 rs61028892-C 标记的第二个单倍型可能包含目前未知的功能等位基因。BCL11A 和 HBS1L-MYB SNP 解释了 24.1%的性状方差。我们的发现为进一步研究镰状细胞病中胎儿血红蛋白持续存在的可变原因提供了途径。