Animal Genomics, ETH Zurich, 8092 Zurich, Switzerland
Animal Genomics, ETH Zurich, 8092 Zurich, Switzerland.
Genome Res. 2024 Mar 20;34(2):300-309. doi: 10.1101/gr.278267.123.
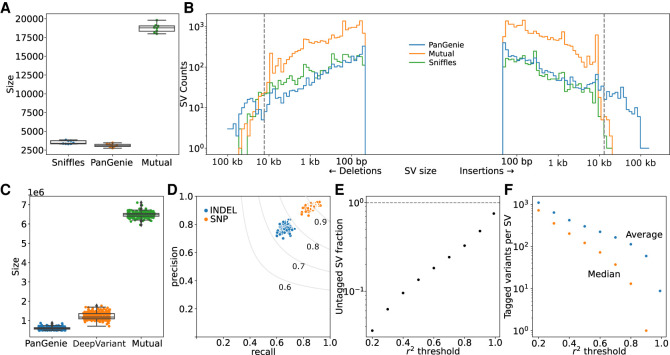
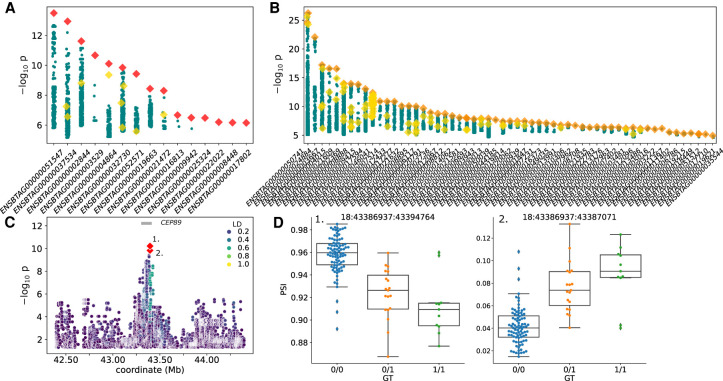
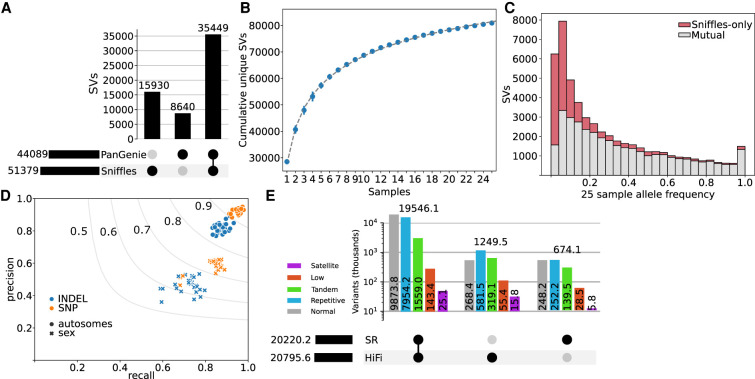
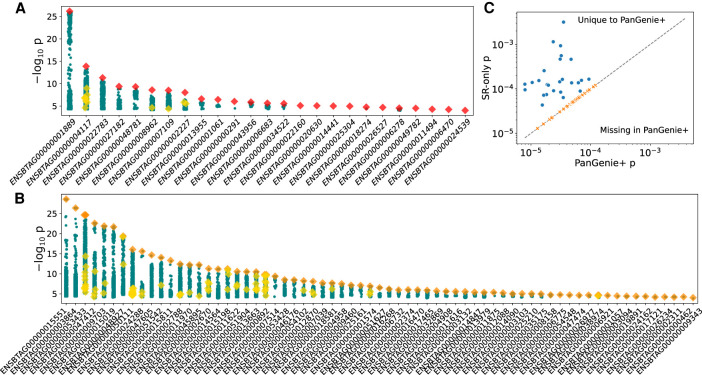
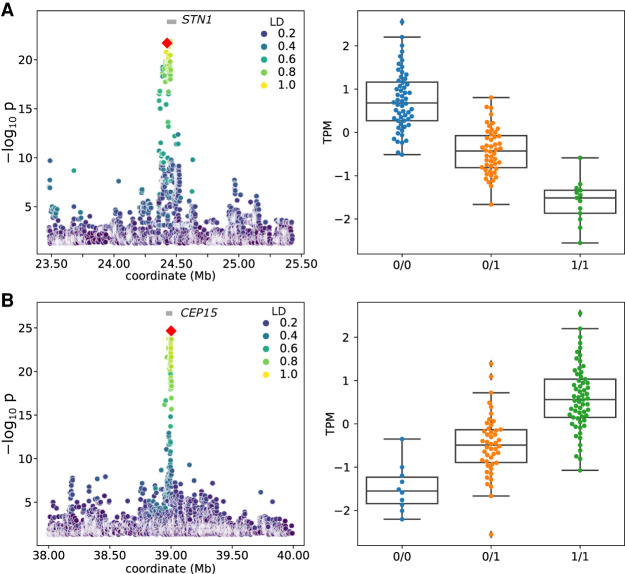
Expression and splicing quantitative trait loci (e/sQTL) are large contributors to phenotypic variability. Achieving sufficient statistical power for e/sQTL mapping requires large cohorts with both genotypes and molecular phenotypes, and so, the genomic variation is often called from short-read alignments, which are unable to comprehensively resolve structural variation. Here we build a pangenome from 16 HiFi haplotype-resolved cattle assemblies to identify small and structural variation and genotype them with PanGenie in 307 short-read samples. We find high (>90%) concordance of PanGenie-genotyped and DeepVariant-called small variation and confidently genotype close to 21 million small and 43,000 structural variants in the larger population. We validate 85% of these structural variants (with MAF > 0.1) directly with a subset of 25 short-read samples that also have medium coverage HiFi reads. We then conduct e/sQTL mapping with this comprehensive variant set in a subset of 117 cattle that have testis transcriptome data, and find 92 structural variants as causal candidates for eQTL and 73 for sQTL. We find that roughly half of the top associated structural variants affecting expression or splicing are transposable elements, such as SV-eQTL for and and SV-sQTL for and Extensive linkage disequilibrium between small and structural variation results in only 28 additional eQTL and 17 sQTL discovered when including SVs, although many top associated SVs are compelling candidates.
表达和剪接数量性状基因座(e/sQTL)是表型变异的主要贡献者。为 e/sQTL 作图实现足够的统计能力需要具有基因型和分子表型的大型队列,因此,基因组变异通常是从短读长比对中调用的,这无法全面解析结构变异。在这里,我们构建了一个来自 16 个 HiFi 单倍型解析牛组装的泛基因组,以识别小和结构变异,并在 307 个短读长样本中使用 PanGenie 对其进行基因分型。我们发现 PanGenie 基因分型和 DeepVariant 调用的小变异的高度(>90%)一致性,并在更大的群体中自信地对近 2100 万个小变异和 43000 个结构变异进行基因分型。我们使用也具有中等覆盖度 HiFi 读数的 25 个短读长样本的子集直接验证了这些结构变异的 85%(MAF > 0.1)。然后,我们在具有睾丸转录组数据的 117 头牛的子集上进行了全面变异集的 e/sQTL 作图,并找到了 92 个结构变异作为 eQTL 的因果候选者和 73 个作为 sQTL 的因果候选者。我们发现,大约一半影响表达或剪接的最相关结构变异是转座元件,例如 上的 SV-eQTL 和 上的 SV-sQTL 和 。小变异和结构变异之间广泛的连锁不平衡导致仅当包含 SVs 时才发现 28 个额外的 eQTL 和 17 个 sQTL,尽管许多顶级关联的 SVs 是令人信服的候选者。