Potter Barney I, Thijssen Marijn, Trovão Nídia Sequeira, Pineda-Peña Andrea, Reynders Marijke, Mina Thomas, Alvarez Carolina, Amini-Bavil-Olyaee Samad, Nevens Frederik, Maes Piet, Lemey Philippe, Van Ranst Marc, Baele Guy, Pourkarim Mahmoud Reza
Department of Microbiology, Immunology and Transplantation, KU Leuven, Rega Institute, Laboratory for Clinical and Epidemiological Virology, Herestraat 49, Leuven BE-3000, Belgium.
Division of International Epidemiology and Population Studies, Fogarty International Center, National Institutes of Health, Bethesda, MD 20892, United States.
Virus Evol. 2024 Feb 1;10(1):veae009. doi: 10.1093/ve/veae009. eCollection 2024.
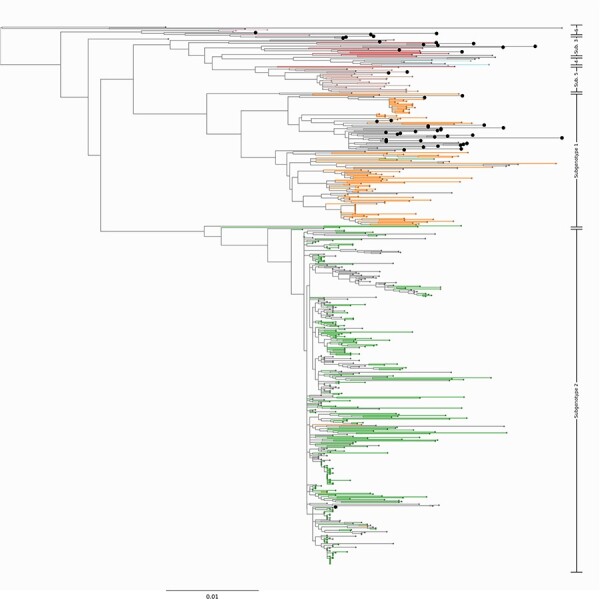
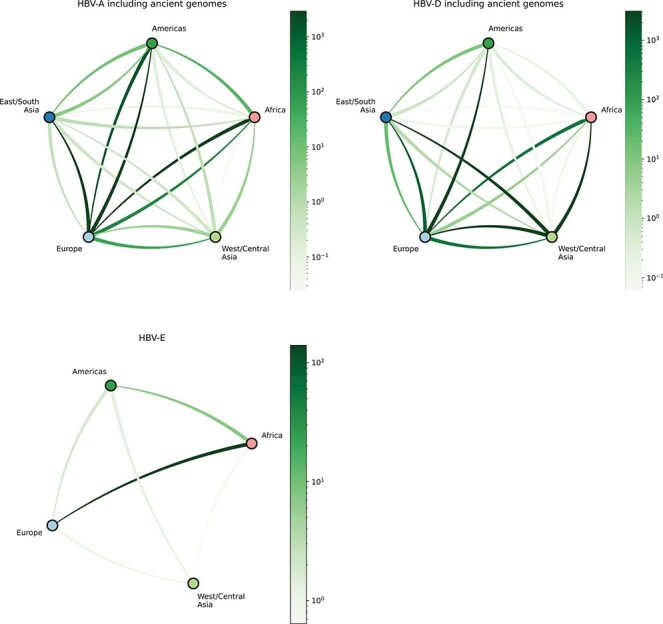
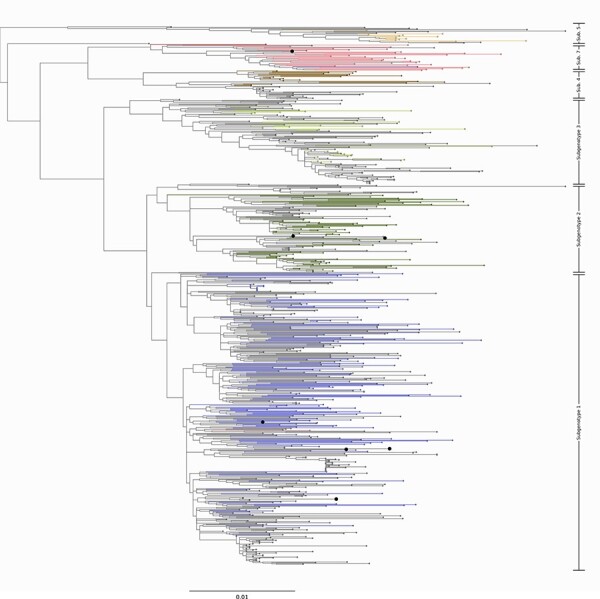
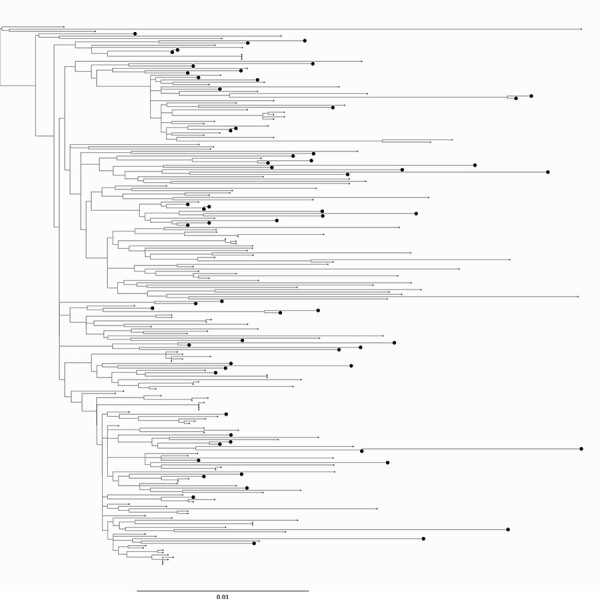
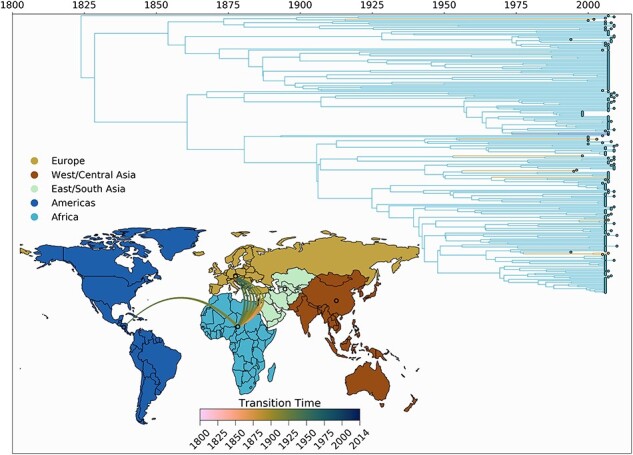
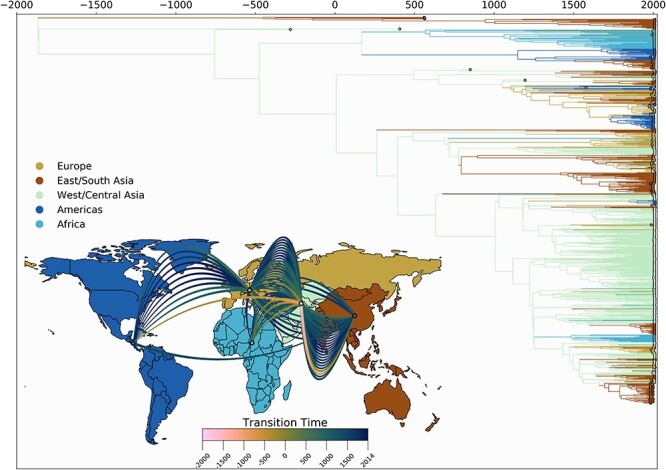
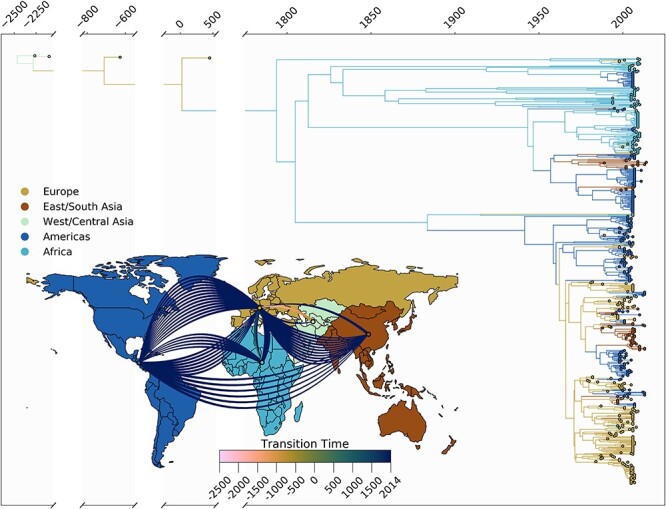
Infection by hepatitis B virus (HBV) is responsible for approximately 296 million chronic cases of hepatitis B, and roughly 880,000 deaths annually. The global burden of HBV is distributed unevenly, largely owing to the heterogeneous geographic distribution of its subtypes, each of which demonstrates different severity and responsiveness to antiviral therapy. It is therefore crucial to the global public health response to HBV that the spatiotemporal spread of each genotype is well characterized. In this study, we describe a collection of 133 newly sequenced HBV strains from recent African immigrants upon their arrival in Belgium. We incorporate these sequences-all of which we determine to come from genotypes A, D, and E-into a large-scale phylogeographic study with genomes sampled across the globe. We focus on investigating the spatio-temporal processes shaping the evolutionary history of the three genotypes we observe. We incorporate several recently published ancient HBV genomes for genotypes A and D to aid our analysis. We show that different spatio-temporal processes underlie the A, D, and E genotypes with the former two having originated in southeastern Asia, after which they spread across the world. The HBV E genotype is estimated to have originated in Africa, after which it spread to Europe and the Americas. Our results highlight the use of phylogeographic reconstruction as a tool to understand the recent spatiotemporal dynamics of HBV, and highlight the importance of supporting vulnerable populations in accordance with the needs presented by specific HBV genotypes.
乙型肝炎病毒(HBV)感染导致约2.96亿例慢性乙型肝炎病例,每年约有88万人死亡。HBV的全球负担分布不均,这主要归因于其亚型在地理上的异质分布,每种亚型对抗病毒治疗的严重程度和反应各不相同。因此,充分了解每种基因型的时空传播情况对于全球应对HBV的公共卫生措施至关重要。在本研究中,我们描述了一组来自近期抵达比利时的非洲移民的133株新测序的HBV毒株。我们将这些序列(所有序列均确定来自A、D和E基因型)纳入一项大规模系统发育地理学研究,该研究涉及全球范围内采集的基因组样本。我们重点研究塑造我们所观察到的三种基因型进化历史的时空过程。我们纳入了一些最近发表的A和D基因型的古代HBV基因组以辅助分析。我们发现,A、D和E基因型有着不同的时空形成过程,前两种基因型起源于东南亚,之后传播到世界各地。据估计,HBV E基因型起源于非洲,之后传播到欧洲和美洲。我们的研究结果凸显了利用系统发育地理学重建作为理解HBV近期时空动态的工具的作用,并强调了根据特定HBV基因型的需求为弱势群体提供支持的重要性。