Department of Orthopaedics, The Fifth Affiliated Hospital of Sun Yat-Sen University, Zhuhai 519000, Guangdong, China.
Department of Trauma Orthopedic, Shenzhen People's Hospital, The Second Clinical Medical College of Jinan University and The First Affiliated Hospital of Southern University of Science and Technology, Shenzhen 518035, Guangdong, China.
Int J Biol Sci. 2024 Jan 27;20(4):1256-1278. doi: 10.7150/ijbs.85585. eCollection 2024.
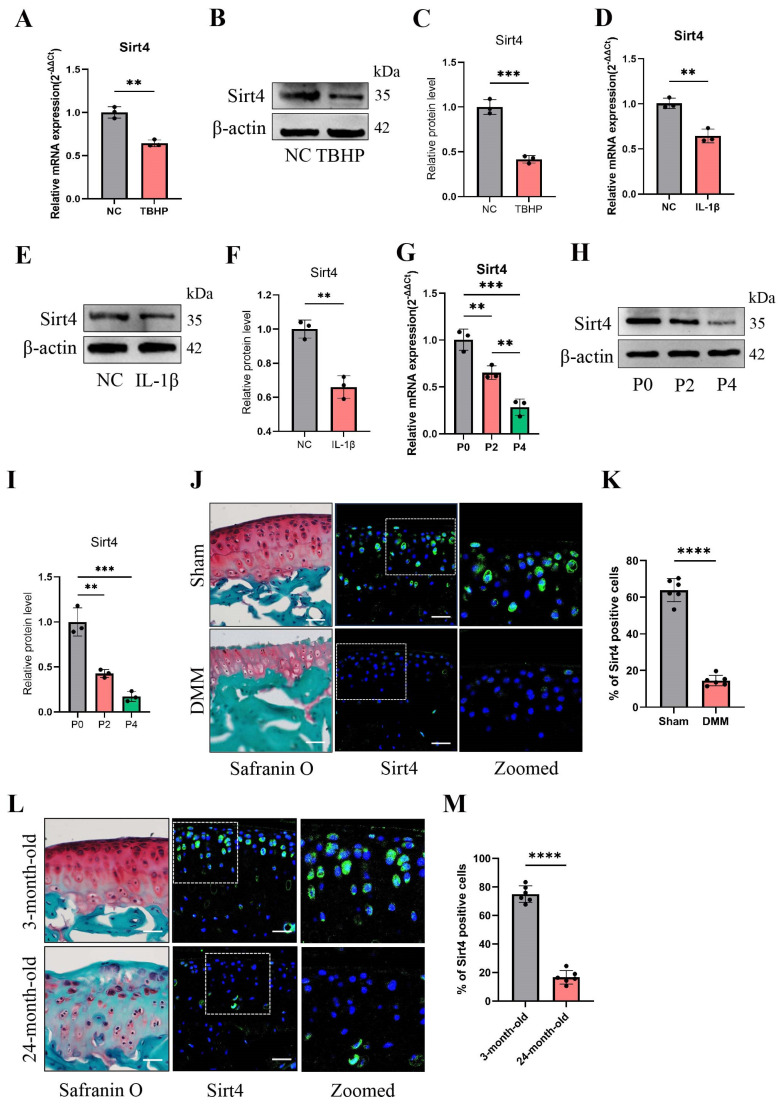
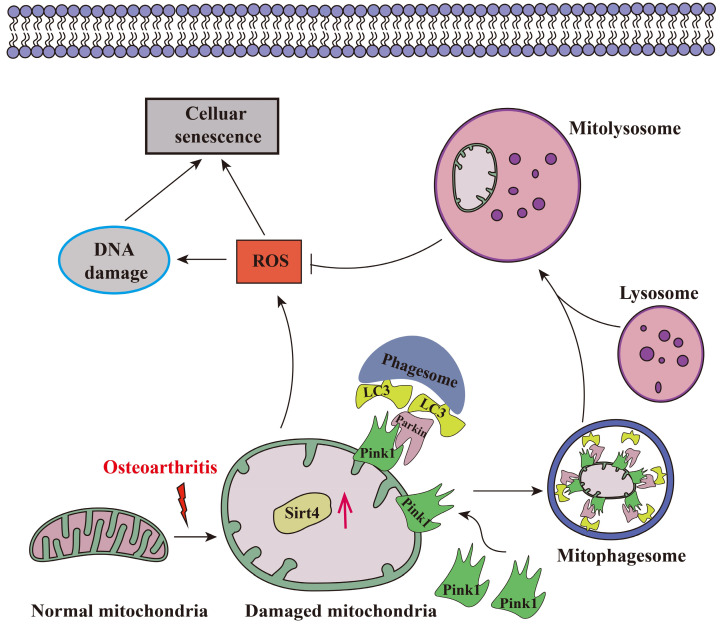
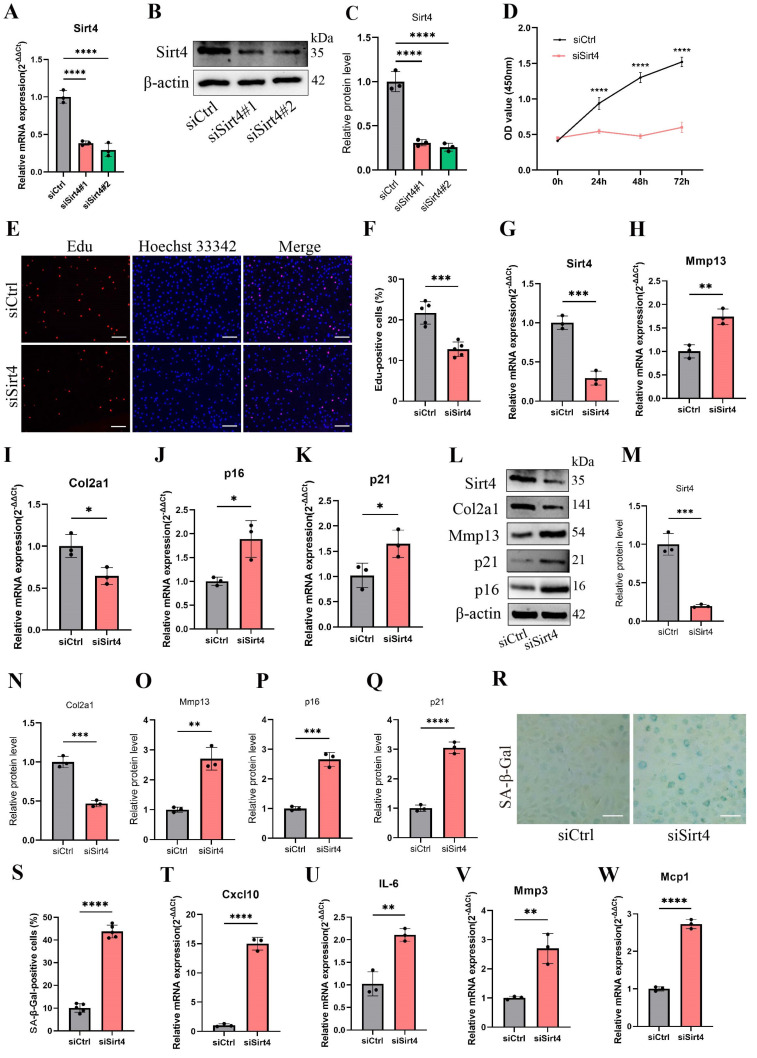
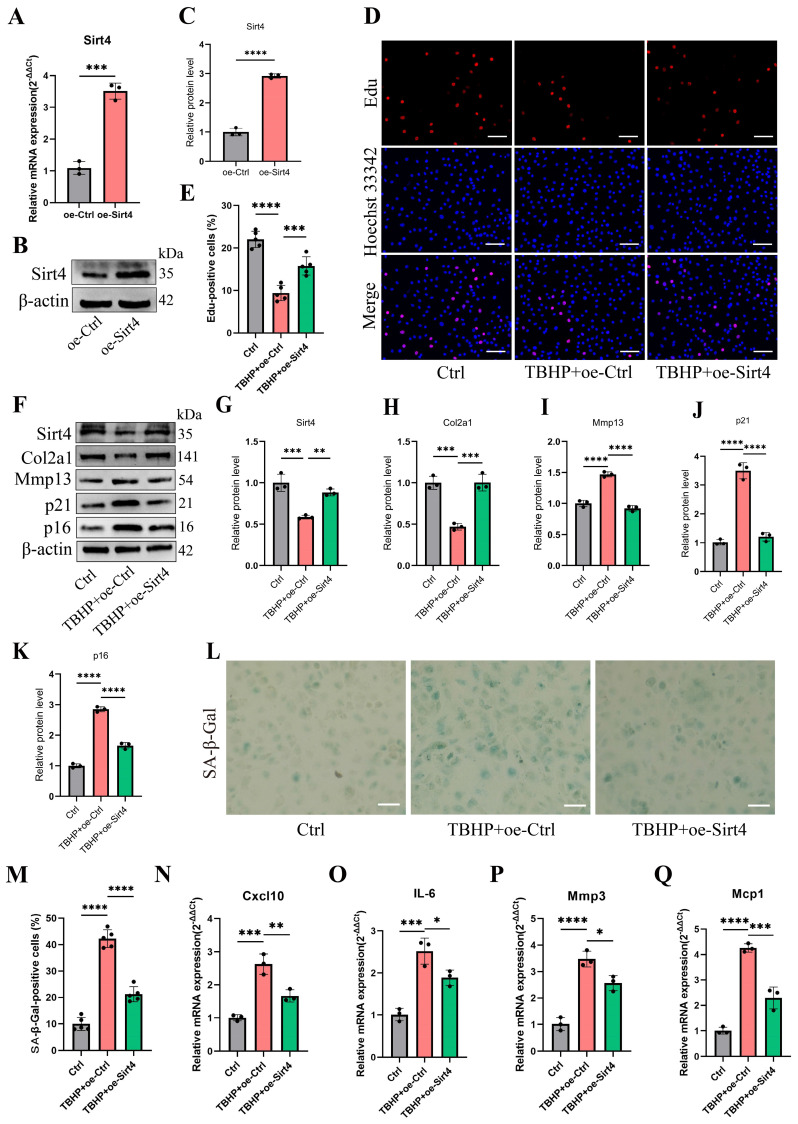
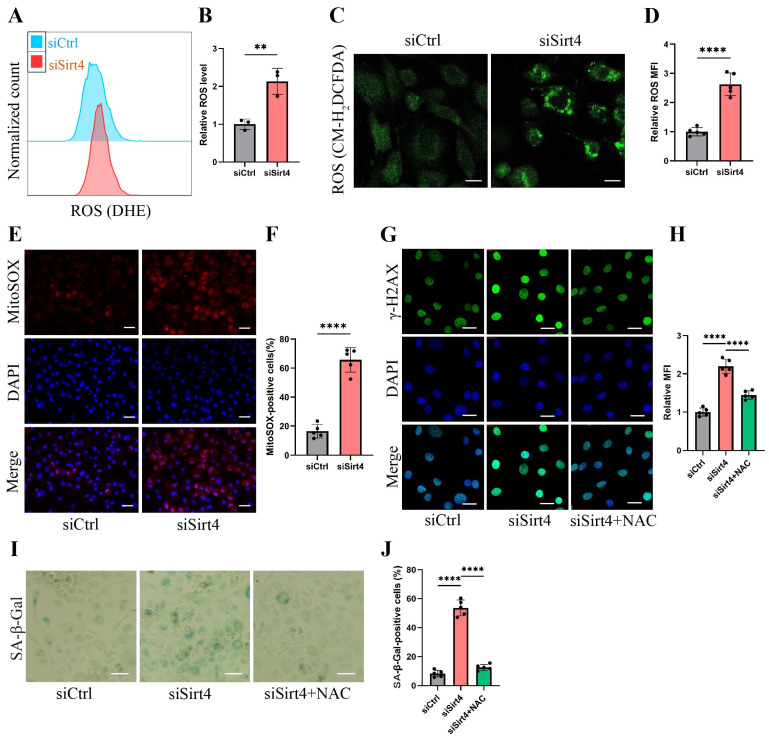
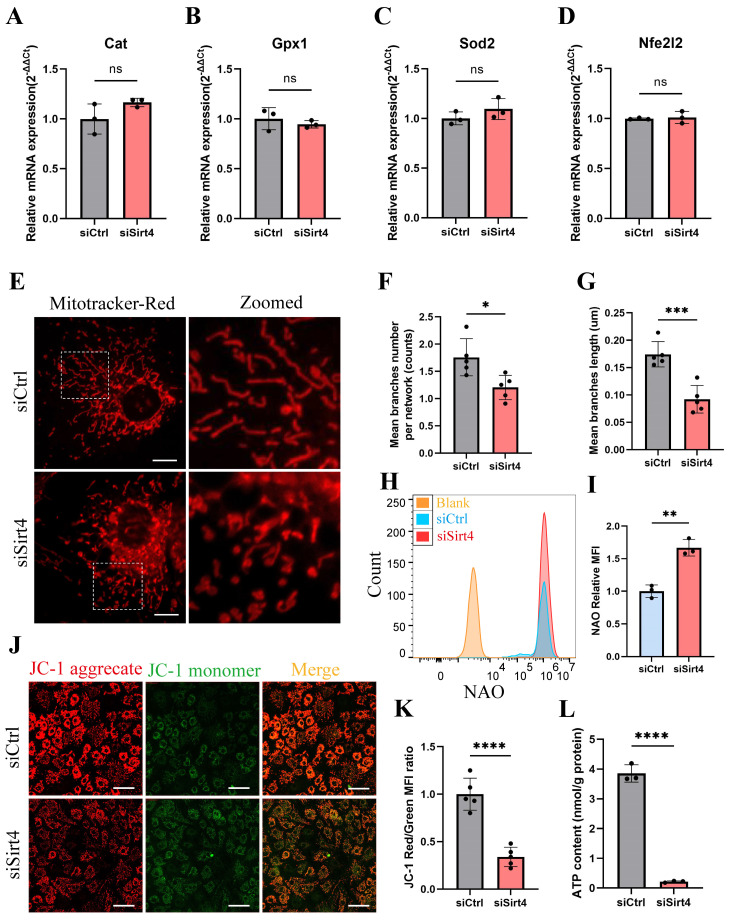
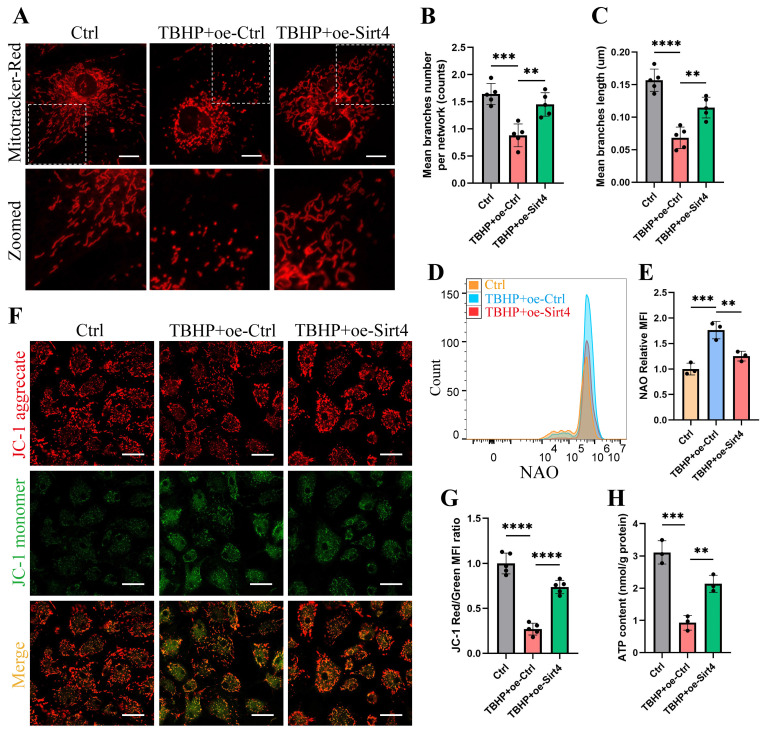
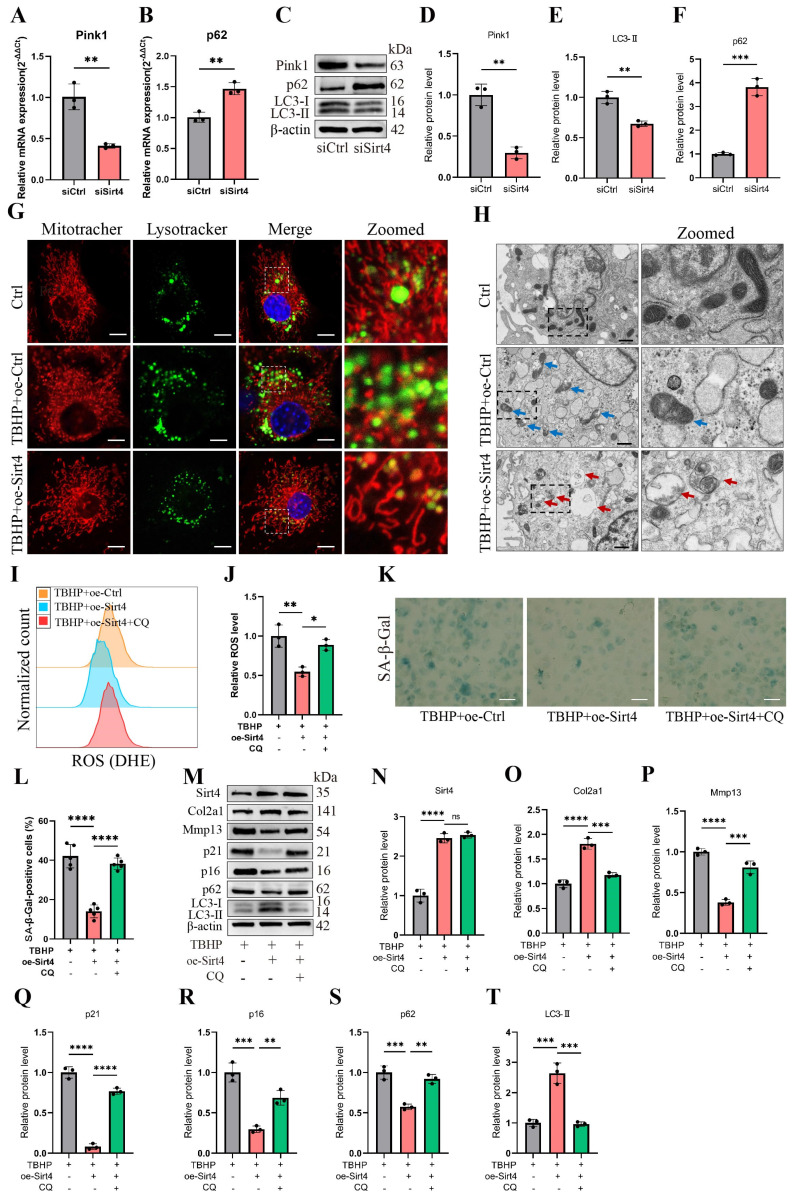
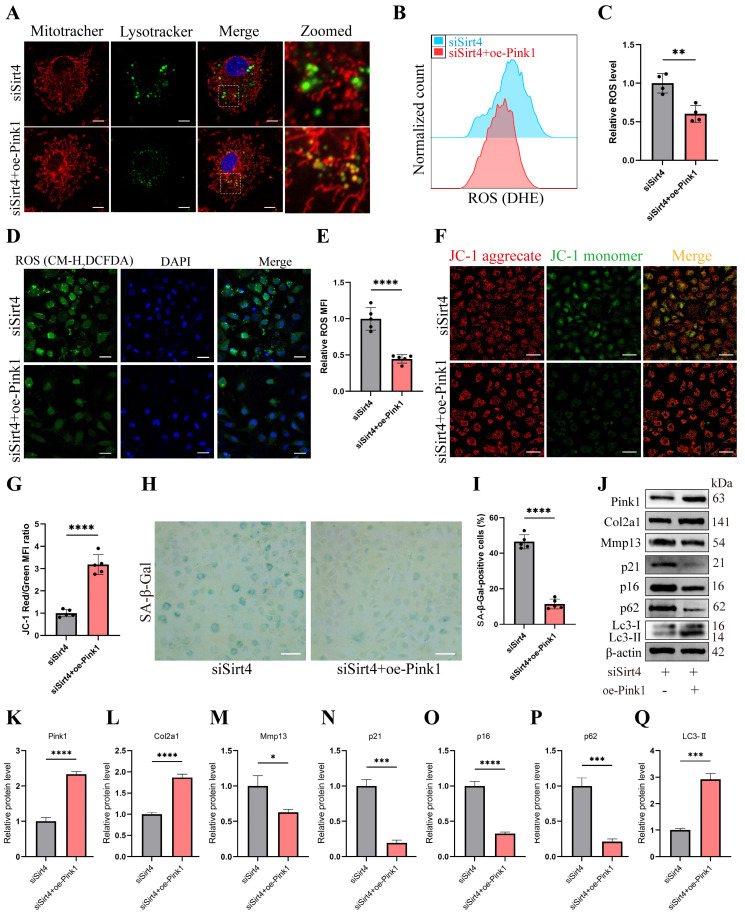
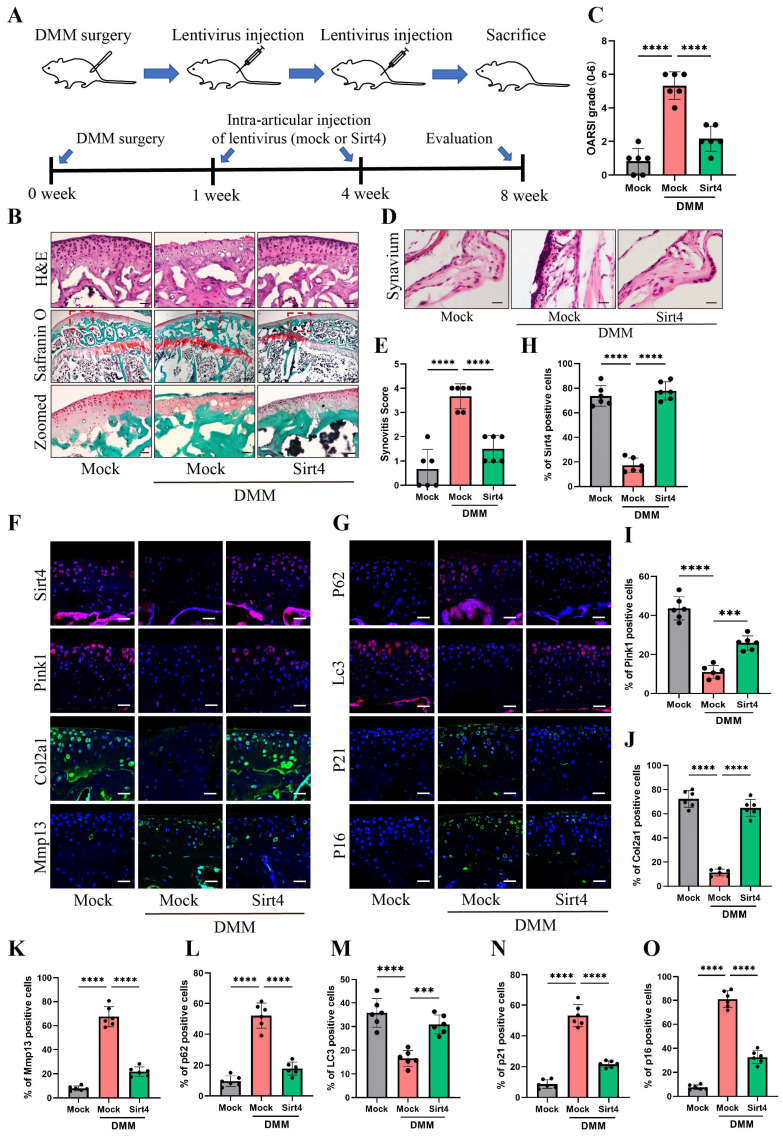
Chondrocyte senescence has recently been proposed as a key pathogenic mechanism in the etiology of osteoarthritis (OA). Nevertheless, the precise molecular mechanisms underlying chondrocyte senescence remain poorly understood. To address this knowledge gap, we conducted an investigation into the involvement of Sirtuin 4 (Sirt4) in chondrocyte senescence. Our experimental findings revealed a downregulation of Sirt4 expression in TBHP-induced senescent chondrocytes in vitro, as well as in mouse OA cartilage. Additionally, we observed that the knockdown of Sirt4 in chondrocytes promoted cellular senescence and cartilage degradation, while the overexpression of Sirt4 protected the cells against TBHP-mediated senescence of chondrocytes and cartilage degradation. Moreover, our findings revealed elevated levels of reactive oxygen species (ROS), abnormal mitochondrial morphology, compromised mitochondrial membrane potential, and reduced ATP production in Sirt4 knockdown chondrocytes, indicative of mitochondrial dysfunction. Conversely, Sirt4 overexpression successfully mitigated TBHP-induced mitochondrial dysfunction. Further analysis revealed that Sirt4 downregulation impaired the cellular capacity to eliminate damaged mitochondria by inhibiting Pink1 in chondrocytes, thereby enhancing the accumulation of ROS and facilitating chondrocyte senescence. Notably, the overexpression of Pink1 counteracted the effects of Sirt4 knockdown on mitochondrial dysfunction. Importantly, our study demonstrated the promise of gene therapy employing a lentiviral vector encoding mouse Sirt4, as it successfully preserved the integrity of articular cartilage in mouse models of OA. In conclusion, our findings provide compelling evidence that the overexpression of Sirt4 enhances mitophagy, restores mitochondrial function, and protects against chondrocyte senescence, thereby offering a novel therapeutic target and potential strategy for the treatment of OA.
软骨细胞衰老最近被提出作为骨关节炎(OA)发病机制的关键致病机制。然而,软骨细胞衰老的确切分子机制仍知之甚少。为了解决这一知识空白,我们研究了 Sirtuin 4(Sirt4)在软骨细胞衰老中的作用。我们的实验结果表明,TBHP 诱导的体外衰老软骨细胞和小鼠 OA 软骨中 Sirt4 表达下调。此外,我们观察到软骨细胞中 Sirt4 的敲低促进了细胞衰老和软骨降解,而过表达 Sirt4 则保护细胞免受 TBHP 介导的软骨细胞衰老和软骨降解。此外,我们的研究结果显示,Sirt4 敲低的软骨细胞中活性氧(ROS)水平升高、线粒体形态异常、线粒体膜电位受损和 ATP 产生减少,表明线粒体功能障碍。相反,Sirt4 的过表达成功缓解了 TBHP 诱导的线粒体功能障碍。进一步分析表明,Sirt4 下调通过抑制 Pink1 损害了软骨细胞清除受损线粒体的能力,从而增强 ROS 的积累并促进软骨细胞衰老。值得注意的是,Pink1 的过表达抵消了 Sirt4 敲低对线粒体功能障碍的影响。重要的是,我们的研究表明,采用编码小鼠 Sirt4 的慢病毒载体进行基因治疗具有很大的潜力,因为它成功地保护了 OA 小鼠模型中关节软骨的完整性。总之,我们的研究结果提供了有力的证据,表明 Sirt4 的过表达增强了线粒体自噬,恢复了线粒体功能,并防止了软骨细胞衰老,从而为 OA 的治疗提供了一个新的治疗靶点和潜在策略。