Tran Van Khanh, Cao My Ha, Nguyen Thi Thanh Hai, Le Phuong Thi, Tran Hai Anh, Vu Dung Chi, Nguyen Ha Thu, Nguyen Mai Thi Phương, Bui The-Hung, Nguyen Thanh Binh, Ta Thanh Van, Tran Thinh Huy
Center for Gene and Protein Research, Hanoi Medical University, Hanoi, Vietnam.
Department of Molecular Pathology, Faculty of Medical Laboratory Technology, Hanoi Medical University, Hanoi, Vietnam.
Front Pediatr. 2024 Feb 13;12:1165492. doi: 10.3389/fped.2024.1165492. eCollection 2024.
Pathogenic variants in the gene are associated with two distinct autosomal recessive neuromuscular disorders: spinal muscular atrophy with respiratory distress type 1 (SMARD1; OMIM #604320) and Charcot-Marie-Tooth type 2S (CMT2S; OMIM #616155). SMARD1 is a severe and fatal condition characterized by infantile-onset respiratory distress, diaphragmatic palsy, and distal muscular weakness, while CMT2S follows a milder clinical course, with slowly progressive distal muscle weakness and sensory loss, without manifestations of respiratory disorder.
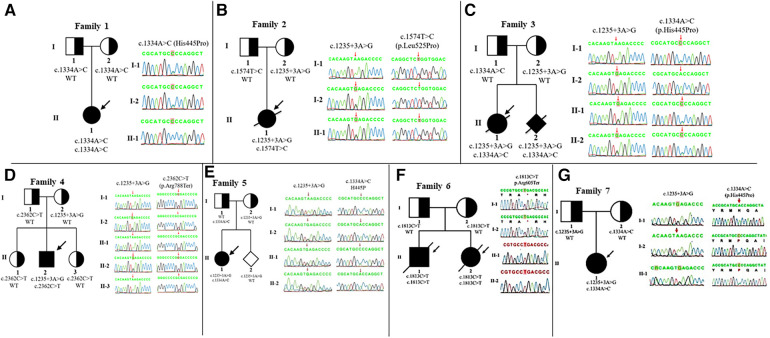
Whole-exome sequencing of the gene was performed for eight Vietnamese patients with -related neuromuscular disorders including five patients with SMARD1 and the others with CMT2S.
We identified one novel variant c.1574T > C (p.Leu525Pro) in a SMARD1 patient. Besides that, two patients shared the same pathogenic variants (c.1235 + 3A > G/c.1334A > C) but presented completely different clinical courses: one with SMARD1 who deceased at 8 months of age, the other with CMT2S was alive at 3 years old without any respiratory distress.
This study is the first to report -related neuromuscular disorders in Vietnam. A novel variant c.1574T > C (p.Leu525Pro) expressing SMARD1 phenotype was detected. The presence of three patients with the same genotype but distinct clinical outcomes suggested the interaction of variants and other factors including relating modified genes in the mechanism of various phenotypes.
该基因的致病变异与两种不同的常染色体隐性神经肌肉疾病相关:1型伴有呼吸窘迫的脊髓性肌萎缩症(SMARD1;OMIM #604320)和2S型夏科-马里-图斯病(CMT2S;OMIM #616155)。SMARD1是一种严重的致命疾病,其特征为婴儿期发作的呼吸窘迫、膈肌麻痹和远端肌肉无力,而CMT2S的临床病程较为温和,表现为远端肌肉无力和感觉丧失缓慢进展,无呼吸障碍表现。
对8名患有与该基因相关神经肌肉疾病的越南患者进行了全外显子组测序,其中包括5名SMARD1患者和其他CMT2S患者。
我们在一名SMARD1患者中鉴定出一种新的基因变异c.1574T>C(p.Leu525Pro)。除此之外,两名患者共享相同的致病变异(c.1235+3A>G/c.1334A>C),但呈现出完全不同的临床病程:一名患有SMARD1的患者在8个月大时死亡,另一名患有CMT2S的患者在3岁时存活,无任何呼吸窘迫症状。
本研究首次报道了越南与该基因相关的神经肌肉疾病。检测到一种表达SMARD1表型的新基因变异c.1574T>C(p.Leu525Pro)。三名基因型相同但临床结果不同的患者的存在表明,变异与其他因素(包括相关修饰基因)在各种表型机制中存在相互作用。