Compain Guillaume, Monsarrat Clément, Blagojevic Julie, Brillet Karl, Dumas Philippe, Hammann Philippe, Kuhn Lauriane, Martiel Isabelle, Engilberge Sylvain, Oliéric Vincent, Wolff Philippe, Burnouf Dominique Y, Wagner Jérôme, Guichard Gilles
Univ. Bordeaux, CNRS, Bordeaux INP, CBMN, UMR 5248, IECB, 2 Rue Robert Escarpit, F-33607 Pessac, France.
Université de Strasbourg, CNRS, FR1589, Plateforme Protéomique Strasbourg Esplanade, 2 Allée K. Roentgen, 67084 Strasbourg, France.
JACS Au. 2024 Jan 25;4(2):432-440. doi: 10.1021/jacsau.3c00572. eCollection 2024 Feb 26.
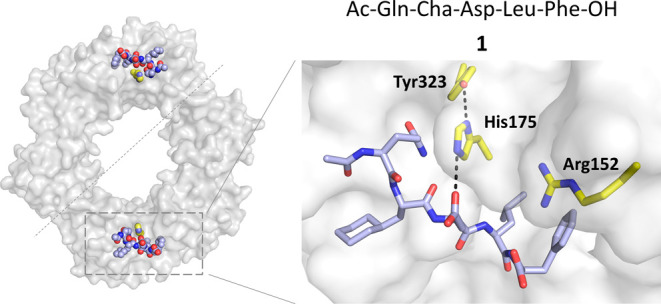
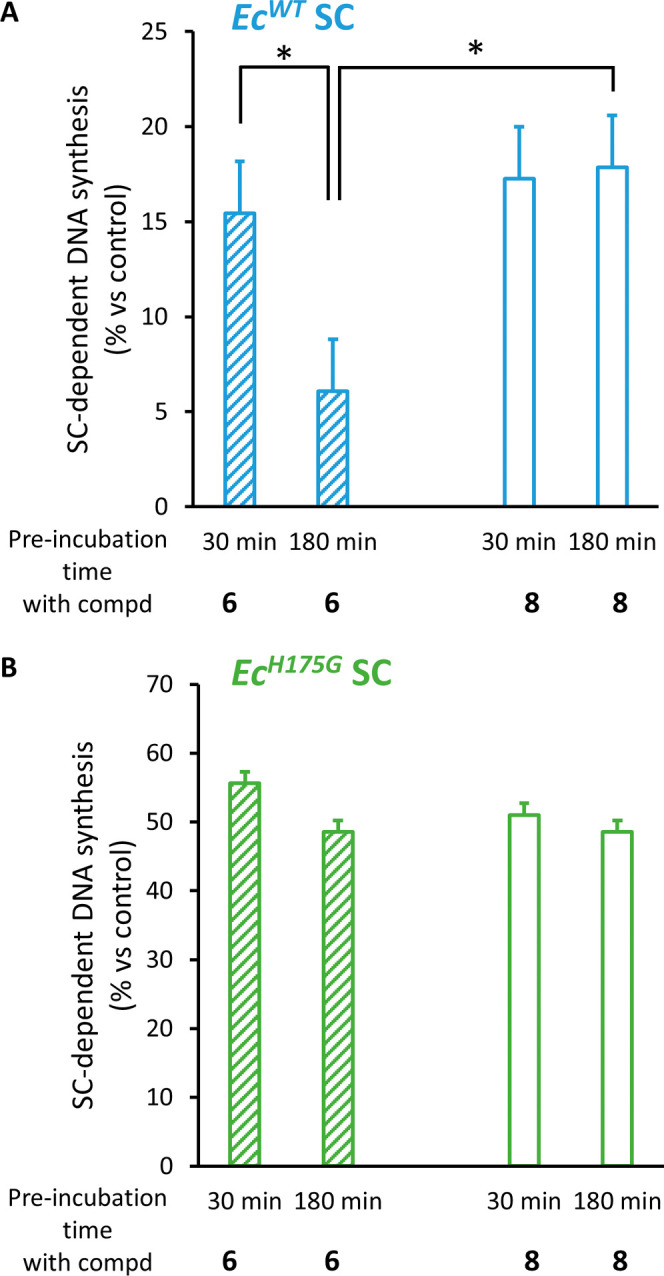
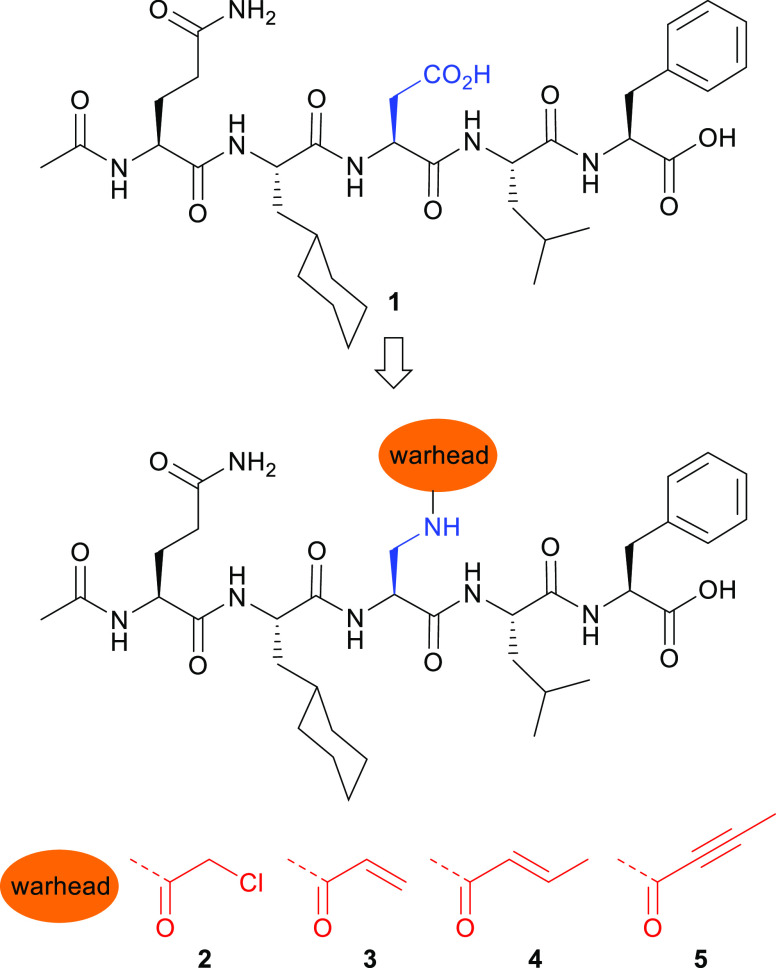
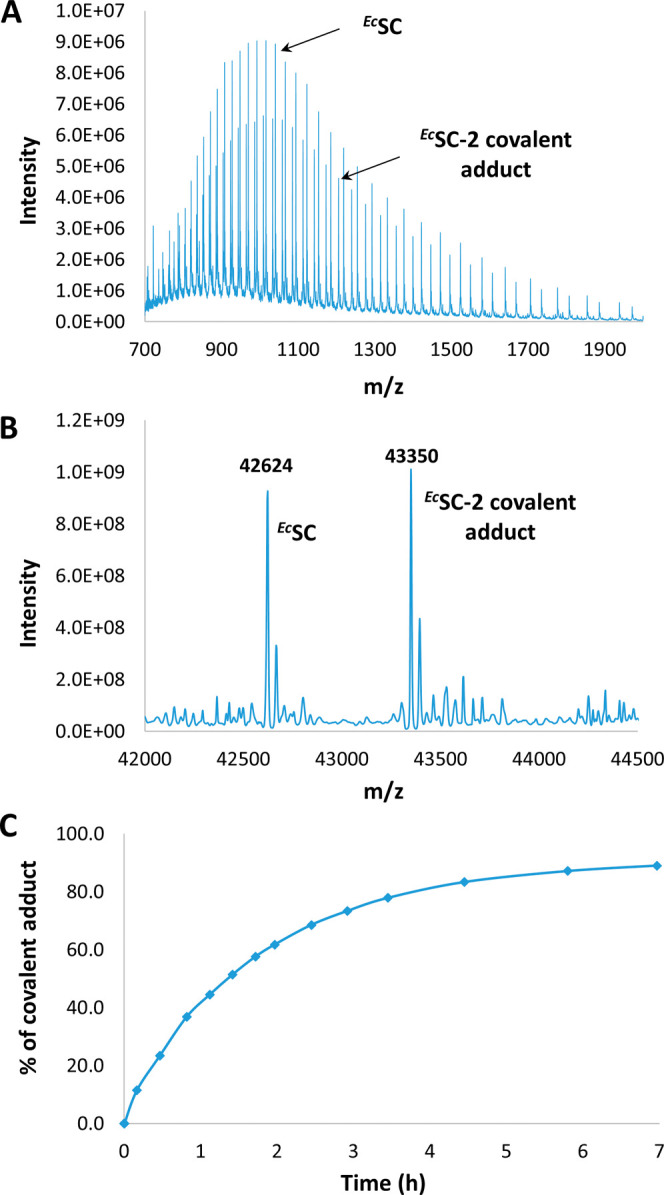
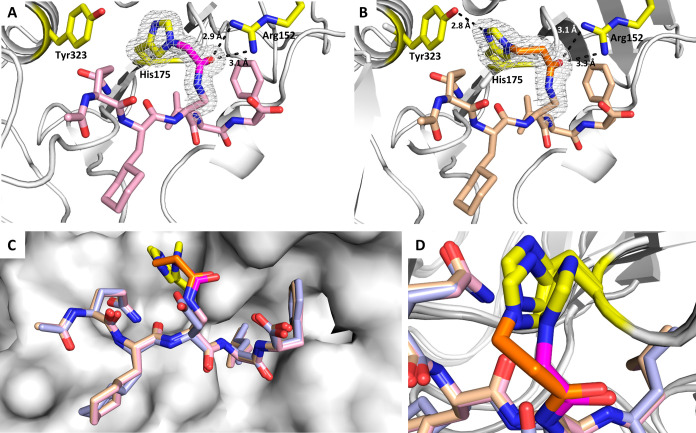
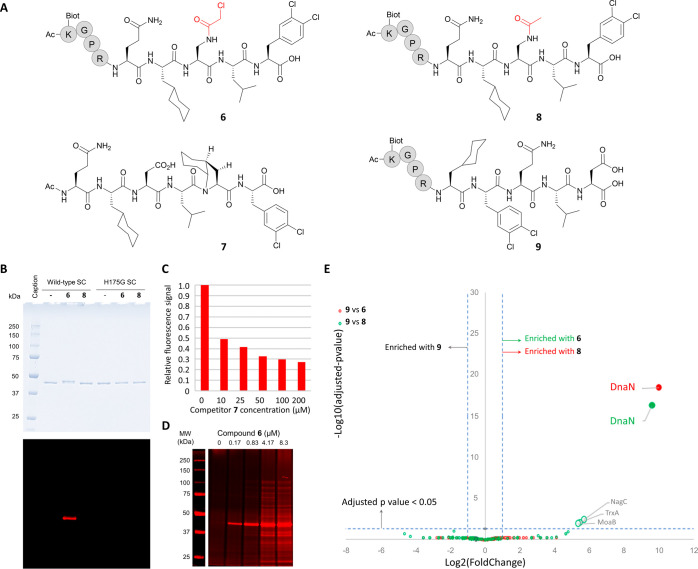
Peptide-based covalent inhibitors targeted to nucleophilic protein residues have recently emerged as new modalities to target protein-protein interactions (PPIs) as they may provide some benefits over more classic competitive inhibitors. Covalent inhibitors are generally targeted to cysteine, the most intrinsically reactive amino acid residue, and to lysine, which is more abundant at the surface of proteins but much less frequently to histidine. Herein, we report the structure-guided design of targeted covalent inhibitors (TCIs) able to bind covalently and selectively to the bacterial sliding clamp (SC), by reacting with a well-conserved histidine residue located on the edge of the peptide-binding pocket. SC is an essential component of the bacterial DNA replication machinery, identified as a promising target for the development of new antibacterial compounds. Thermodynamic and kinetic analyses of ligands bearing different mild electrophilic warheads confirmed the higher efficiency of the chloroacetamide compared to Michael acceptors. Two high-resolution X-ray structures of covalent inhibitor-SC adducts were obtained, revealing the canonical orientation of the ligand and details of covalent bond formation with histidine. Proteomic studies were consistent with a selective SC engagement by the chloroacetamide-based TCI. Finally, the TCI of SC was substantially more active than the parent noncovalent inhibitor in an in vitro SC-dependent DNA synthesis assay, validating the potential of the approach to design covalent inhibitors of protein-protein interactions targeted to histidine.
靶向亲核蛋白残基的肽基共价抑制剂最近作为一种靶向蛋白质-蛋白质相互作用(PPI)的新方法出现,因为它们可能比更经典的竞争性抑制剂具有一些优势。共价抑制剂通常靶向半胱氨酸(最具内在反应性的氨基酸残基)和赖氨酸(在蛋白质表面更丰富,但与组氨酸的结合频率低得多)。在此,我们报告了靶向共价抑制剂(TCI)的结构导向设计,该抑制剂能够通过与位于肽结合口袋边缘的一个保守组氨酸残基反应,与细菌滑动夹(SC)共价且选择性地结合。SC是细菌DNA复制机制的一个重要组成部分,被确定为开发新型抗菌化合物的一个有前景的靶点。对带有不同温和亲电弹头的配体进行的热力学和动力学分析证实,氯乙酰胺比迈克尔受体具有更高的效率。获得了共价抑制剂-SC加合物的两个高分辨率X射线结构,揭示了配体的标准取向以及与组氨酸形成共价键的细节。蛋白质组学研究与基于氯乙酰胺的TCI对SC的选择性结合一致。最后,在体外SC依赖性DNA合成试验中,SC的TCI比母体非共价抑制剂活性高得多,验证了设计靶向组氨酸的蛋白质-蛋白质相互作用共价抑制剂方法的潜力。