The Morris Kahn Laboratory of Human Genetics at the National Institute of Biotechnology in the Negev and Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel.
The Shraga Segal Department of Microbiology, Immunology, and Genetics, Faculty of Health Science, Ben-Gurion University of the Negev, Beer Sheva, Israel.
Eur J Hum Genet. 2024 May;32(5):550-557. doi: 10.1038/s41431-024-01559-1. Epub 2024 Mar 4.
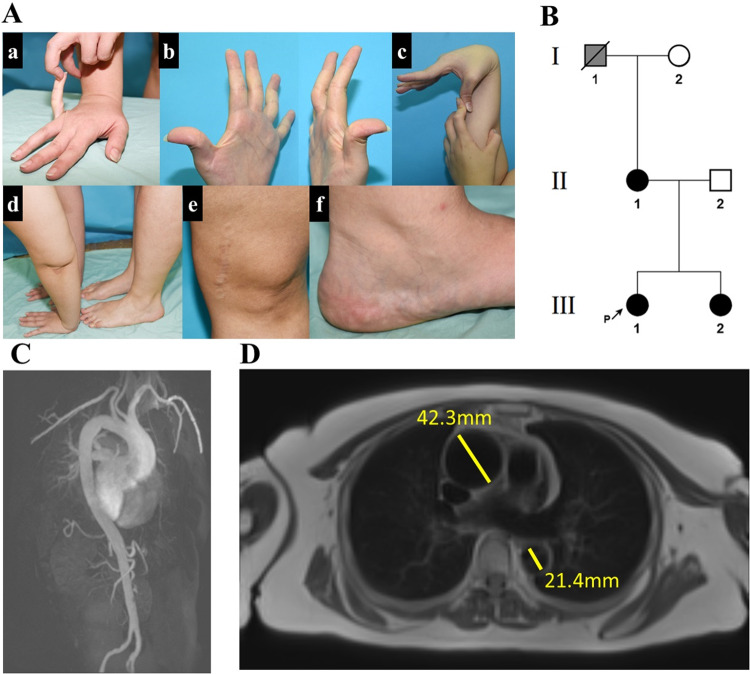
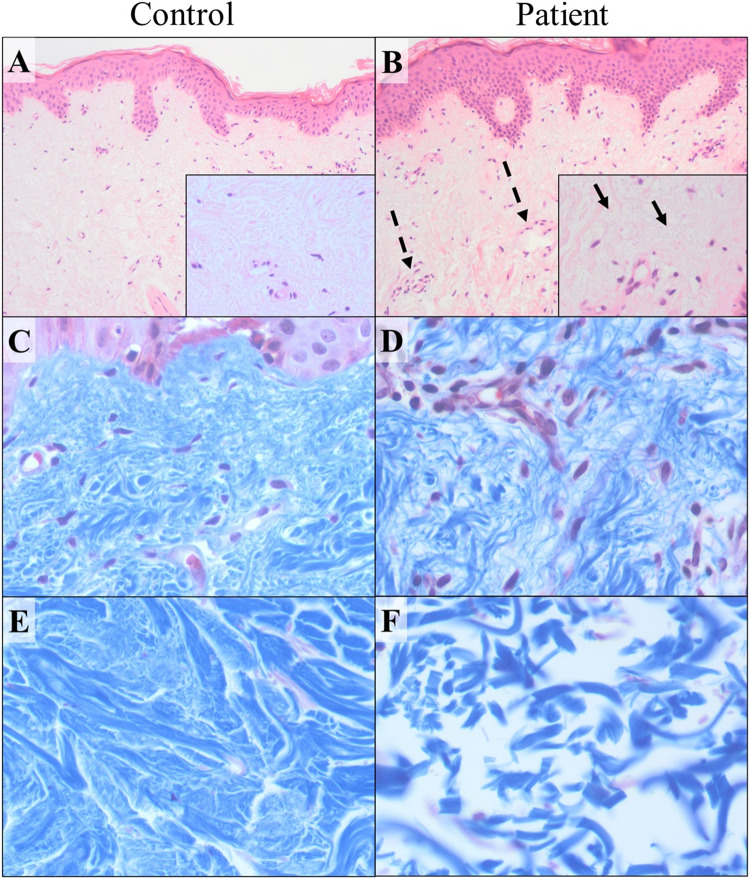
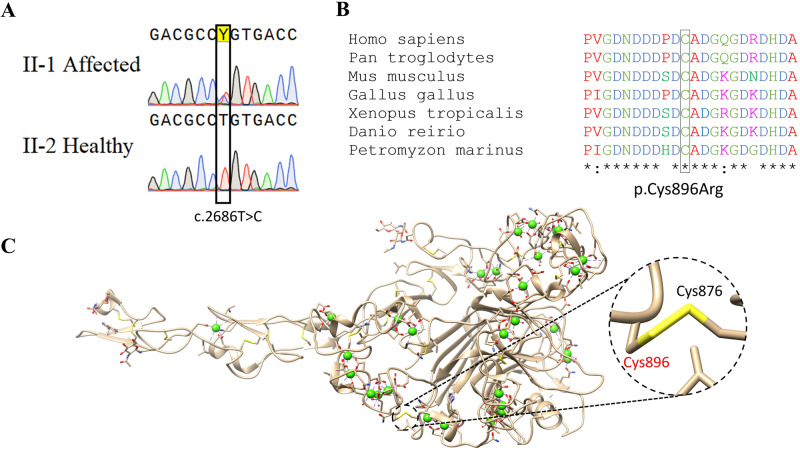
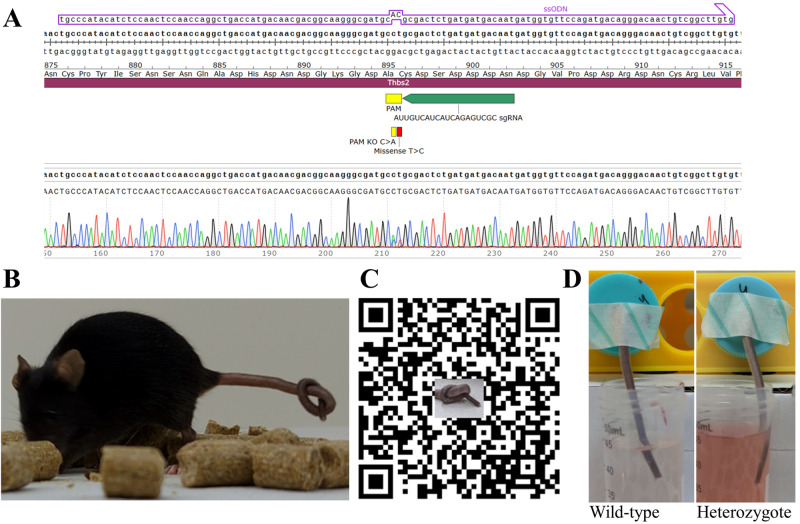
Ehlers-Danlos syndromes (EDS) are a group of connective tissue disorders caused by mutations in collagen and collagen-interacting genes. We delineate a novel form of EDS with vascular features through clinical and histopathological phenotyping and genetic studies of a three-generation pedigree, displaying an apparently autosomal dominant phenotype of joint hypermobility and frequent joint dislocations, atrophic scarring, prolonged bleeding time and age-related aortic dilatation and rupture. Coagulation tests as well as platelet counts and function were normal. Reticular dermis displayed highly disorganized collagen fibers and transmission electron microscopy (TEM) revealed abnormally shaped fibroblasts and endothelial cells, with high amount and irregular shape of extracellular matrix (ECM) substance, especially near blood vessels. Genetic analysis unraveled a heterozygous mutation in THBS2 (NM_003247.5:c.2686T>C, p.Cys896Arg). We generated CRISPR/Cas9 knock-in (KI) mice, bearing the heterozygous human mutation in the mouse ortholog. The KI mice demonstrated phenotypic traits correlating with those observed in the human subjects, as evidenced by morphologic, histologic, and TEM analyses, in conjunction with bleeding time assays. Our findings delineate a novel form of human EDS with classical-like elements combined with vascular features, caused by a heterozygous THBS2 missense mutation. We further demonstrate a similar phenotype in heterozygous THBS2 KI mice, in line with previous studies in Thbs2 homozygous null-mutant mice. Notably, THBS2 encodes Thrombospondin-2, a secreted homotrimeric matricellular protein that directly binds the ECM-shaping Matrix Metalloproteinase 2 (MMP2), mediating its clearance. THBS2 loss-of-function attenuates MMP2 clearance, enhancing MMP2-mediated proteoglycan cleavage, causing ECM abnormalities similar to those seen in the human and mouse disease we describe.
埃勒斯-当洛斯综合征(EDS)是一组由胶原蛋白和胶原蛋白相互作用基因的突变引起的结缔组织疾病。我们通过对一个三代家系的临床和组织病理学表型及遗传研究,描绘了一种具有血管特征的新型 EDS,其表现为关节过度活动和频繁关节脱位、萎缩性瘢痕、延长的出血时间以及与年龄相关的主动脉扩张和破裂的明显常染色体显性表型。凝血试验以及血小板计数和功能均正常。网状真皮显示高度紊乱的胶原纤维,透射电子显微镜(TEM)显示异常形状的成纤维细胞和内皮细胞,细胞外基质(ECM)物质含量高且形状不规则,尤其是在血管附近。遗传分析揭示了 THBS2 中的杂合突变(NM_003247.5:c.2686T>C,p.Cys896Arg)。我们生成了携带小鼠同源物中杂合人突变的 CRISPR/Cas9 敲入(KI)小鼠。KI 小鼠表现出与人类受试者观察到的表型特征相关的表型特征,这通过形态学、组织学和 TEM 分析以及出血时间测定得到证实。我们的研究结果描绘了一种具有经典样特征与血管特征相结合的新型人类 EDS,其由 THBS2 错义突变引起。我们进一步在杂合 THBS2 KI 小鼠中证明了类似的表型,这与之前在 Thbs2 纯合突变型小鼠中的研究一致。值得注意的是,THBS2 编码 Thrombospondin-2,这是一种分泌的三聚体细胞外基质蛋白,可直接结合 ECM 塑造的基质金属蛋白酶 2(MMP2),介导其清除。THBS2 功能丧失会减弱 MMP2 的清除,增强 MMP2 介导的蛋白聚糖裂解,导致 ECM 异常,类似于我们描述的人类和小鼠疾病中所见。