Zhu Shuyao, Hu Qi, Yang Yunxia, Zhu Hui, Wang Jin, Luo Zemin, Ou Mincai, Chen Ai, Huang Yu, Xiong Fu, Zhou Jiaji, Liu Jinglin, Lei Xunming, Zeng Lan
Department of Pediatrics, Sichuan Provincial Maternity and Child Health Care Hospital, Chengdu, Sichuan, China.
Department of Neonatal Screening Centre in Sichuan Province Maternity and Child Health Care Hospital, Chengdu, Sichuan, China.
Heliyon. 2024 Feb 24;10(5):e27050. doi: 10.1016/j.heliyon.2024.e27050. eCollection 2024 Mar 15.
Tetrahydrobiopterin (BH4) deficiency is a rare cause of hyperphenylalaninemia (HPA). The incidence of this condition varies based on region and ethnicity. In the early stages, patients typically do not exhibit any symptoms, and HPA is identified only through newborn screening for diseases. It is important to distinguish BH4 deficiency from phenylketonuria (PKU, MIM # 261600). Timely diagnosis and treatment of BH4 deficiency are crucial for the prognosis of patients.
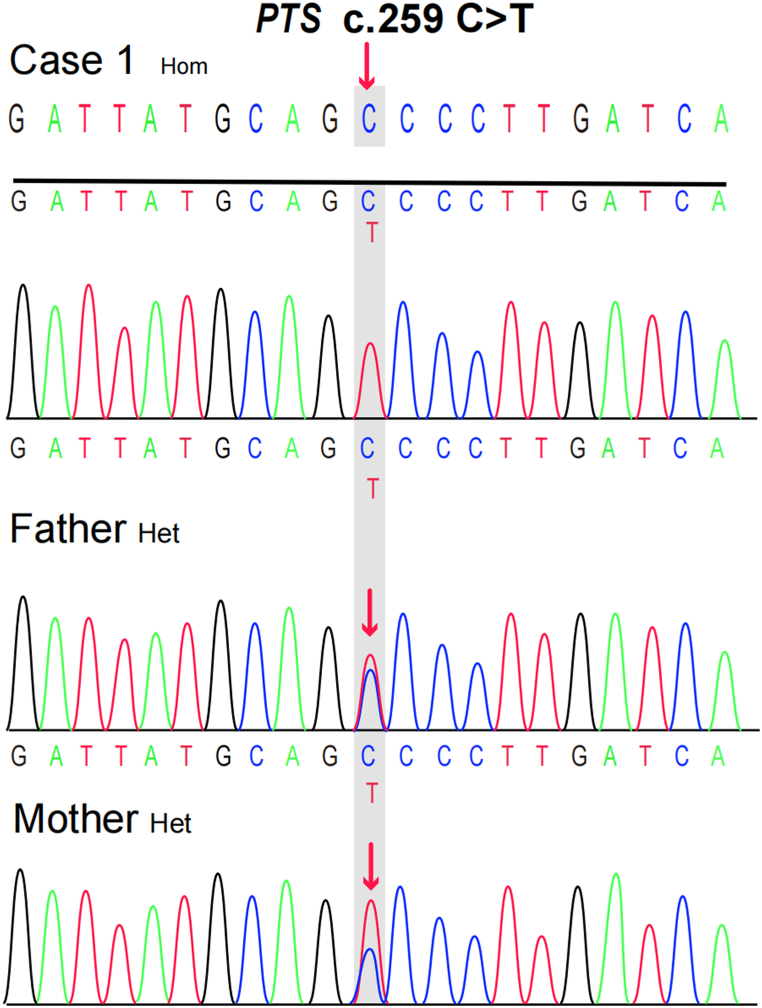
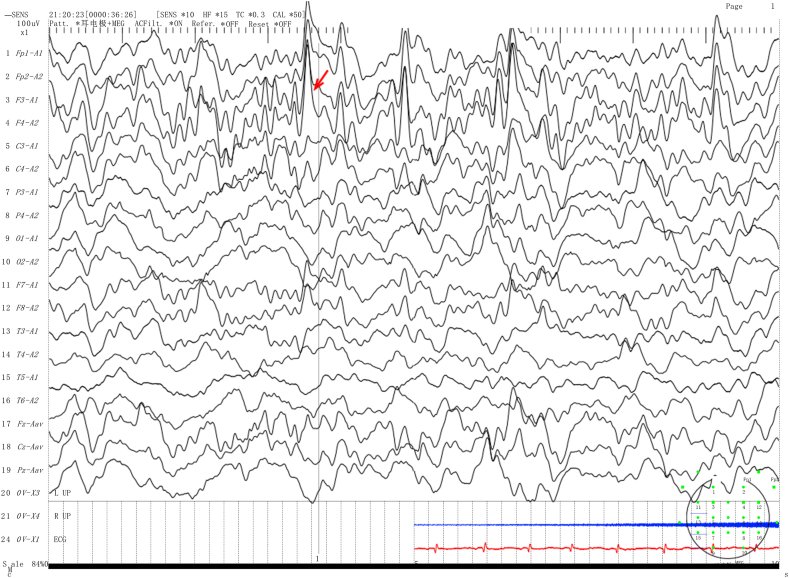
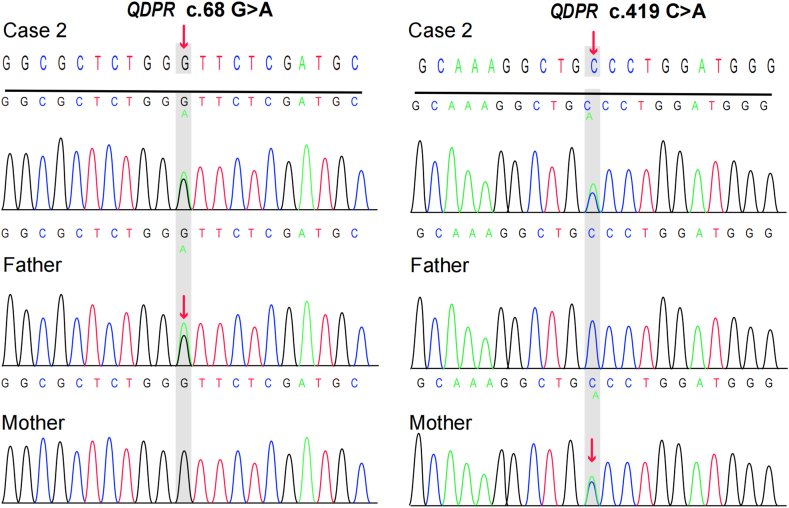
We present two rare cases of Chinese Tibetan children with BH4D, diagnosed through biochemical tests and genetic sequencing. Case 1 is a male infant, 2 months old, with a newborn screening (NBS) Phe level of 1212 μmol/L (reference range <120 μmol). The biopterin(B) level was 0.19 mmol/molCr (reference range: 0.42-1.92 mmol/molCr), with a B% of 5.67% (reference range: 19.8%-50.3%). Gene sequencing revealed a homozygous missense variant [NM_000317.3 (): c.259C > T (p.Pro87Ser), rs104894276, ClinVar variation ID: 480]. The patient was treated with a Phe-reduced diet and oral sapropterin, madopar and is currently 3 years and 4 months old, showing mild global developmental delay. Case 2 is a 40-day-old female infant with a Phe level of 2442.11 μmol/L and dihydropteridine reductase (DHPR) activity of 0.84 nmol/(min. 5 mm disc) (reference range: 1.02-3.35 nmol/min.5 mm disc. Gene sequencing revealed a compound heterozygous genotype [NM_000320.3(): c.68G > A (p.Gly23Asp), rs104893863, ClinVar Variation ID: 490] and [NM_000320.3() c.419C > A (p. Ala140Asp), ClinVar ID: 2444501]. The patient was treated with a Phe-reduced diet and oral madopar, 5-hydroxytryptophan. At the age of 1 year, she exhibited severe global developmental delay with seizures.
We identified and treated two cases of BH4D in Tibetan populations in China, marking the first confirmed instances. Our report emphasizes the significance of conducting differential diagnosis tests for BH4D.
四氢生物蝶呤(BH4)缺乏症是高苯丙氨酸血症(HPA)的罕见病因。该病的发病率因地区和种族而异。在疾病早期,患者通常没有任何症状,HPA仅通过新生儿疾病筛查得以确诊。将BH4缺乏症与苯丙酮尿症(PKU,MIM # 261600)区分开来很重要。及时诊断和治疗BH4缺乏症对患者的预后至关重要。
我们报告了两例中国藏族儿童BH4缺乏症的罕见病例,通过生化检测和基因测序确诊。病例1为一名2个月大的男婴,新生儿筛查(NBS)苯丙氨酸(Phe)水平为1212 μmol/L(参考范围<120 μmol)。生物蝶呤(B)水平为0.19 mmol/mol肌酐(参考范围:0.42 - 1.92 mmol/mol肌酐),B%为5.67%(参考范围:19.8% - 50.3%)。基因测序显示为纯合错义变异[NM_000317.3():c.259C>T(p.Pro87Ser),rs104894276,ClinVar变异ID:480]。该患者接受了低苯丙氨酸饮食以及口服沙丙蝶呤、美多芭治疗,目前3岁4个月,有轻度全面发育迟缓。病例2为一名40天大的女婴,Phe水平为2442.11 μmol/L,二氢蝶啶还原酶(DHPR)活性为0.84 nmol/(min·5 mm圆盘)(参考范围:1.02 - 3.35 nmol/min·5 mm圆盘)。基因测序显示为复合杂合基因型[NM_000320.3():c.68G>A(p.Gly23Asp),rs104893863,ClinVar变异ID:490]和[NM_000320.3()c.419C>A(p.Ala140Asp),ClinVar ID:2444501]。该患者接受了低苯丙氨酸饮食以及口服美多芭、5 - 羟色氨酸治疗。1岁时,她出现了严重的全面发育迟缓并伴有癫痫发作。
我们在中国藏族人群中识别并治疗了两例BH4缺乏症病例,这是首次确诊的病例。我们的报告强调了对BH4缺乏症进行鉴别诊断检测的重要性。