Division of Pediatric Neurology and Developmental Neuroscience, Department of Pediatrics, Baylor College of Medicine, Houston, TX, 77030, USA.
Texas Children's Hospital, Houston, TX, 77030, USA.
BMC Neurol. 2024 Mar 4;24(1):87. doi: 10.1186/s12883-024-03571-w.
RARS2-related mitochondrial disorder is an autosomal recessive mitochondrial encephalopathy caused by biallelic pathogenic variants in the gene encoding the mitochondrial arginyl-transfer RNA synthetase 2 (RARS2, MIM *611524, NM_020320.5). RARS2 catalyzes the transfer of L-arginine to its cognate tRNA during the translation of mitochondrially-encoded proteins. The classical presentation of RARS2-related mitochondrial disorder includes pontocerebellar hypoplasia (PCH), progressive microcephaly, profound developmental delay, feeding difficulties, and hypotonia. Most patients also develop severe epilepsy by three months of age, which consists of focal or generalized seizures that frequently become pharmacoresistant and lead to developmental and epileptic encephalopathy (DEE).
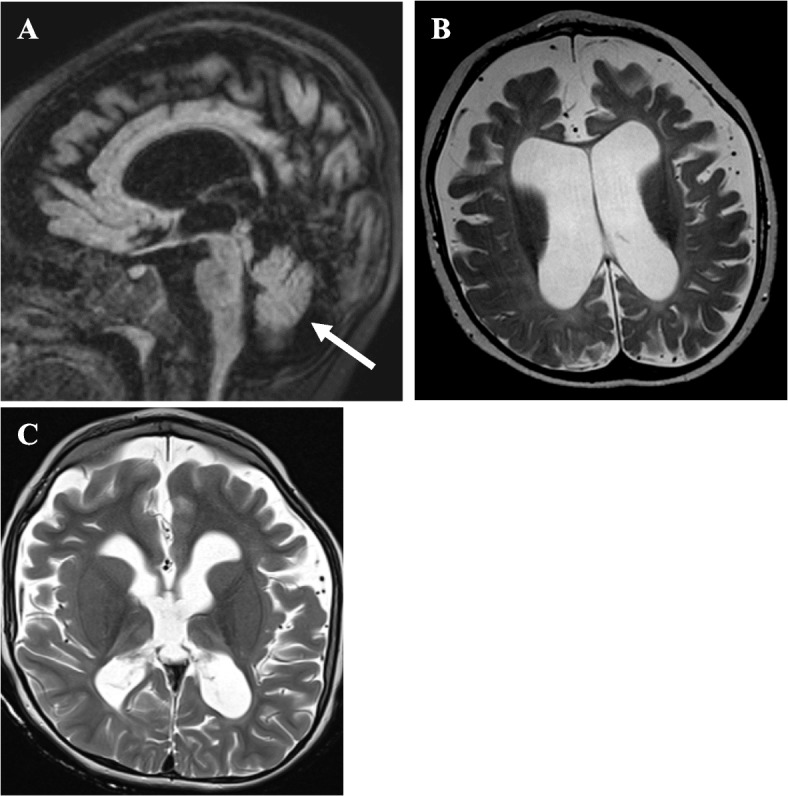
Here, we describe a six-year-old boy with developmental delay, hypotonia, and failure to thrive who developed an early-onset DEE consistent with Lennox-Gastaut Syndrome (LGS), which has not previously been observed in this disorder. He had dysmorphic features including bilateral macrotia, overriding second toes, a depressed nasal bridge, retrognathia, and downslanting palpebral fissures, and he did not demonstrate progressive microcephaly. Whole genome sequencing identified two variants in RARS2, c.36 + 1G > T, a previously unpublished variant that is predicted to affect splicing and is, therefore, likely pathogenic and c.419 T > G (p.Phe140Cys), a known pathogenic variant. He exhibited significant, progressive generalized brain atrophy and ex vacuo dilation of the supratentorial ventricular system on brain MRI and did not demonstrate PCH. Treatment with a ketogenic diet (KD) reduced seizure frequency and enabled him to make developmental progress. Plasma untargeted metabolomics analysis showed increased levels of lysophospholipid and sphingomyelin-related metabolites.
Our work expands the clinical spectrum of RARS2-related mitochondrial disorder, demonstrating that patients can present with dysmorphic features and an absence of progressive microcephaly, which can help guide the diagnosis of this condition. Our case highlights the importance of appropriate seizure phenotyping in this condition and indicates that patients can develop LGS, for which a KD may be a viable therapeutic option. Our work further suggests that analytes of phospholipid metabolism may serve as biomarkers of mitochondrial dysfunction.
RARS2 相关的线粒体疾病是一种常染色体隐性遗传的线粒体脑病,由编码线粒体精氨酰-tRNA 合成酶 2(RARS2,MIM *611524,NM_020320.5)的基因中的双等位致病性变异引起。RARS2 在翻译线粒体编码的蛋白质时催化 L-精氨酸转移到其对应的 tRNA 上。RARS2 相关线粒体疾病的经典表现包括桥脑小脑发育不良(PCH)、进行性小头畸形、严重发育迟缓、喂养困难和肌张力低下。大多数患者在三个月大时也会出现严重的癫痫发作,表现为局灶性或全身性癫痫发作,这些发作往往对药物治疗耐药,并导致发育性和癫痫性脑病(DEE)。
这里,我们描述了一名六岁男孩,他患有发育迟缓、肌张力低下和生长不良,出现了早发性 DEE,与 Lennox-Gastaut 综合征(LGS)一致,这种情况以前在这种疾病中没有观察到。他具有双侧大耳、第二脚趾重叠、鼻梁凹陷、小下颌和眼睑裂下斜等发育不良特征,且没有进行性小头畸形。全基因组测序发现 RARS2 中的两个变异,c.36+1G>T,这是一个以前未发表的变异,据预测会影响剪接,因此很可能是致病性的,以及 c.419T>G(p.Phe140Cys),这是一个已知的致病性变异。他表现出明显的、进行性的全脑萎缩和脑 MRI 上的脑室系统外空性扩张,没有表现出 PCH。酮饮食(KD)治疗降低了癫痫发作的频率,并使他能够取得发育进展。血浆非靶向代谢组学分析显示,溶血磷脂和鞘磷脂相关代谢物的水平升高。
我们的工作扩展了 RARS2 相关线粒体疾病的临床谱,表明患者可能表现出发育不良特征和无进行性小头畸形,这有助于指导该疾病的诊断。我们的病例强调了在这种情况下进行适当的癫痫发作表型分析的重要性,并表明患者可能会出现 LGS,KD 可能是一种可行的治疗选择。我们的工作进一步表明,磷脂代谢的分析物可以作为线粒体功能障碍的生物标志物。