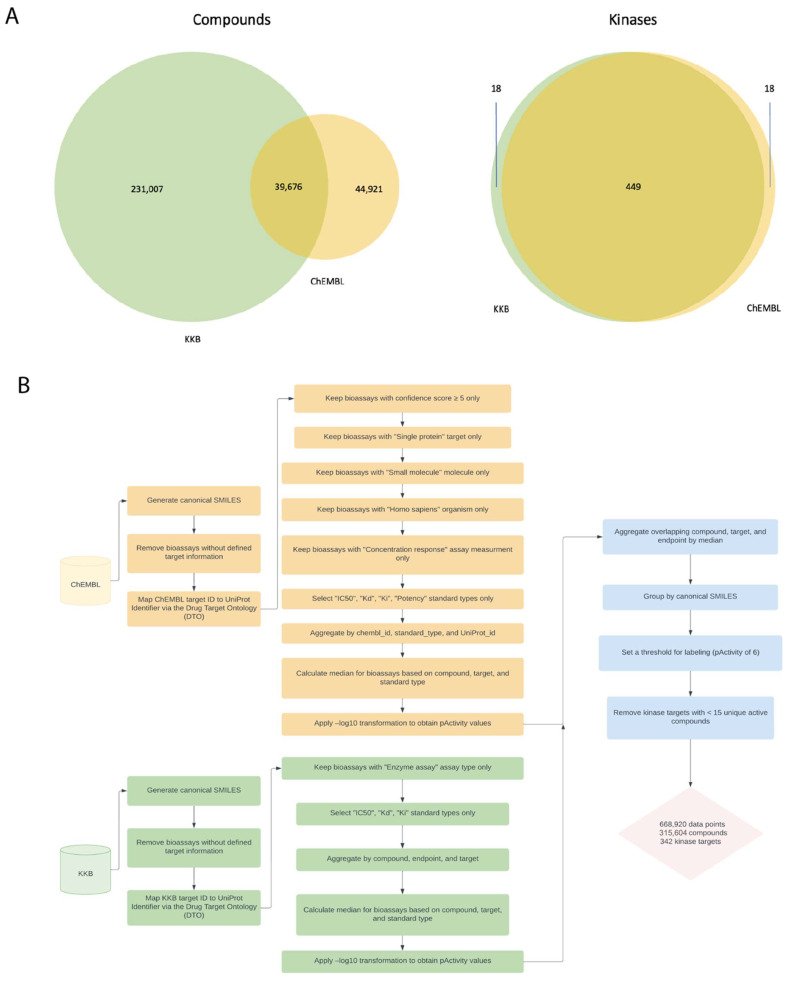
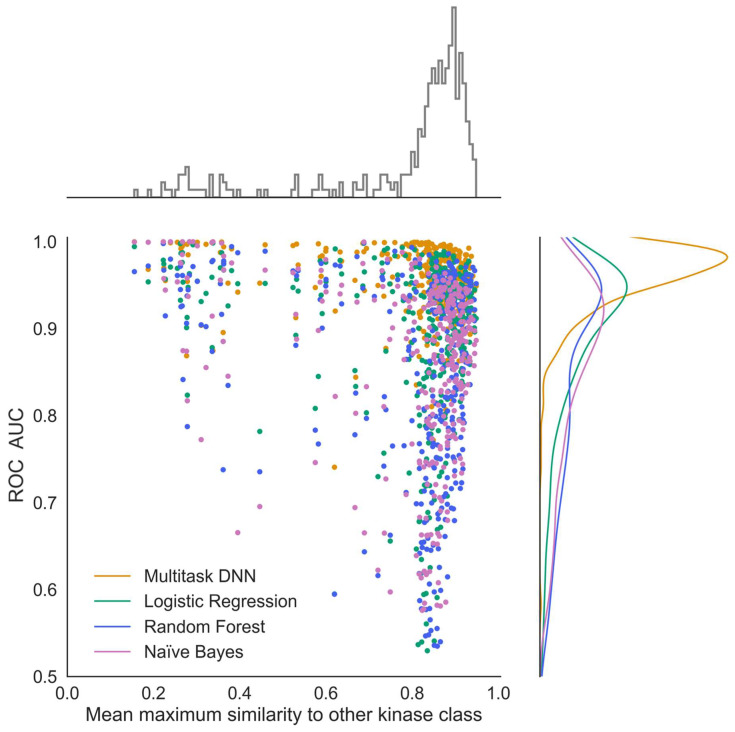
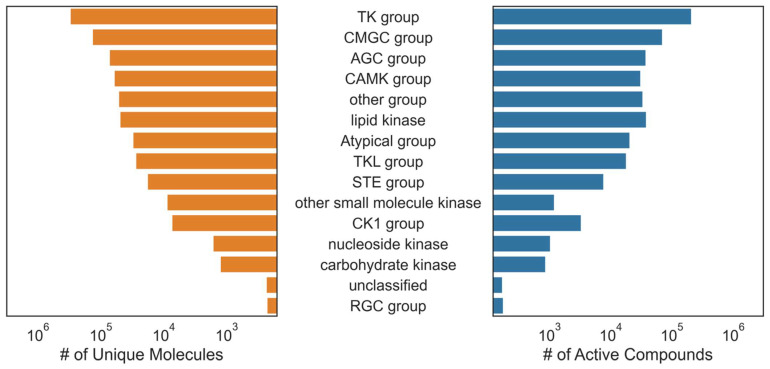
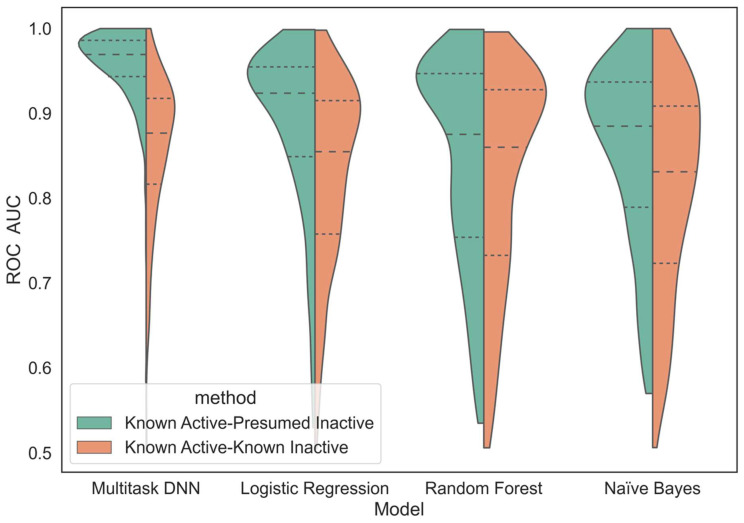
Hu Jiaming, Allen Bryce K, Stathias Vasileios, Ayad Nagi G, Schürer Stephan C
Dr. John T. Macdonald Foundation Department of Human Genetics and John P. Hussman Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, FL 33136, USA.
Department of Molecular and Cellular Pharmacology, Miller School of Medicine, University of Miami, Miami, FL 33136, USA.
Int J Mol Sci. 2024 Feb 22;25(5):2538. doi: 10.3390/ijms25052538.
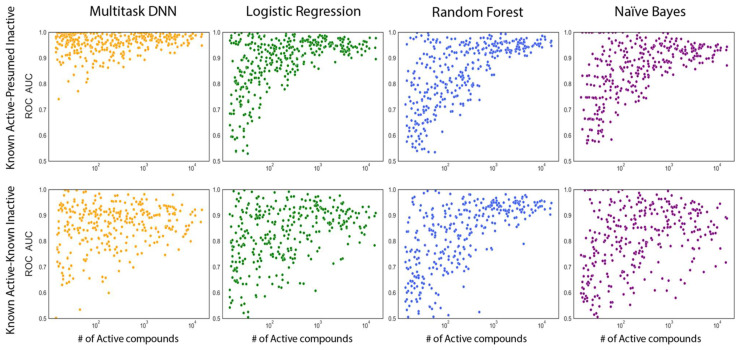
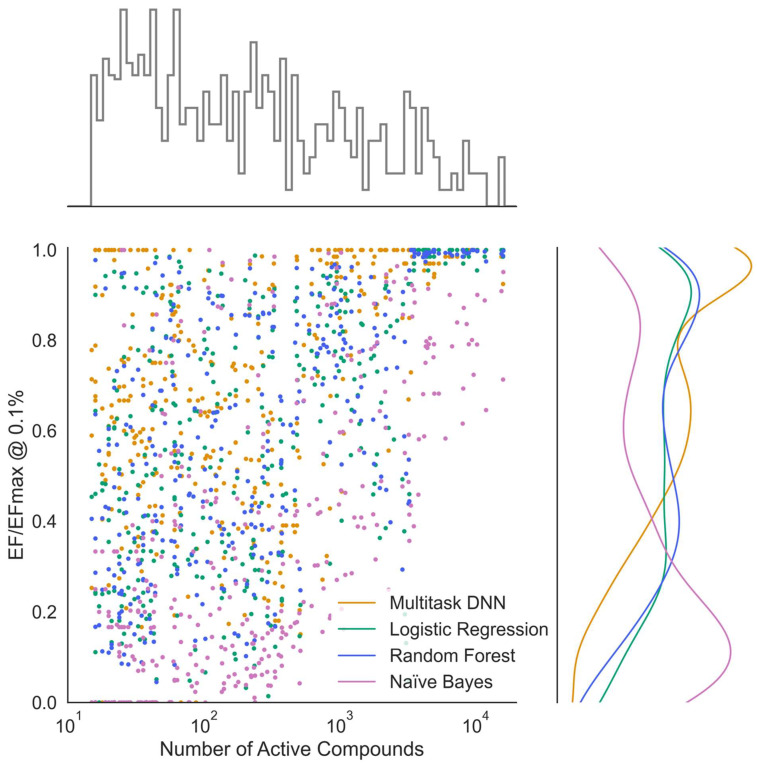
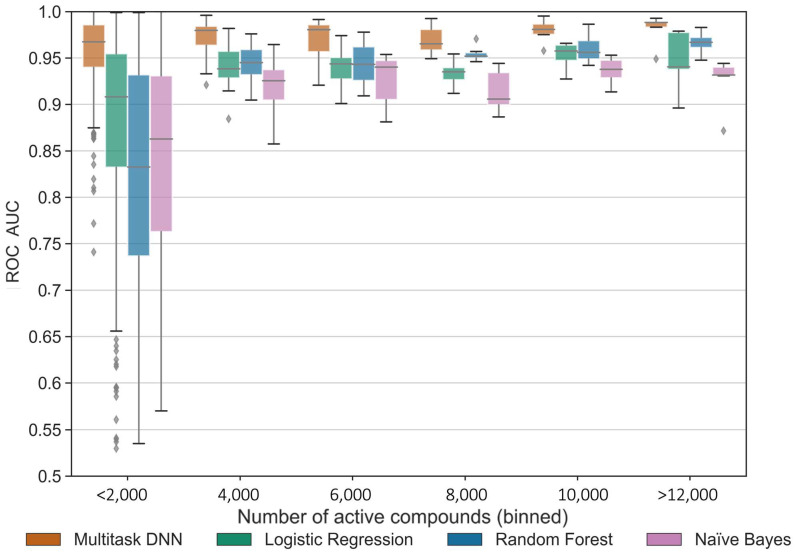
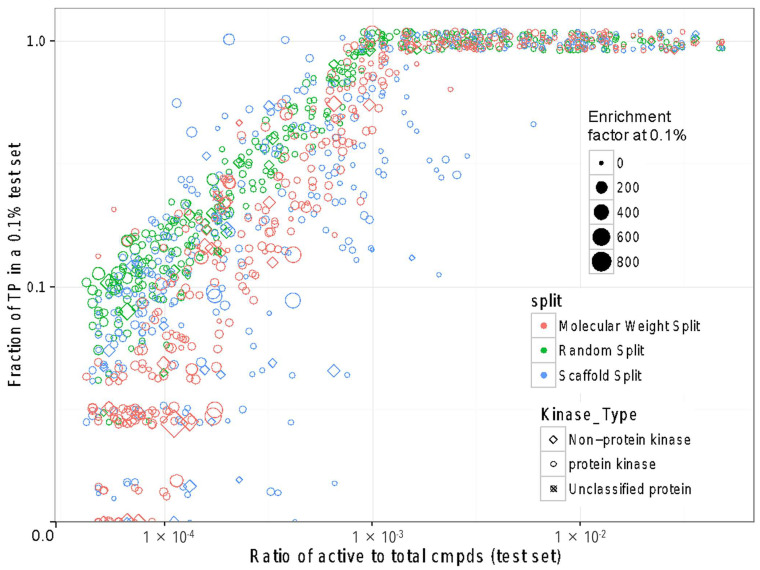
Deep learning is a machine learning technique to model high-level abstractions in data by utilizing a graph composed of multiple processing layers that experience various linear and non-linear transformations. This technique has been shown to perform well for applications in drug discovery, utilizing structural features of small molecules to predict activity. Here, we report a large-scale study to predict the activity of small molecules across the human kinome-a major family of drug targets, particularly in anti-cancer agents. While small-molecule kinase inhibitors exhibit impressive clinical efficacy in several different diseases, resistance often arises through adaptive kinome reprogramming or subpopulation diversity. Polypharmacology and combination therapies offer potential therapeutic strategies for patients with resistant diseases. Their development would benefit from a more comprehensive and dense knowledge of small-molecule inhibition across the human kinome. Leveraging over 650,000 bioactivity annotations for more than 300,000 small molecules, we evaluated multiple machine learning methods to predict the small-molecule inhibition of 342 kinases across the human kinome. Our results demonstrated that multi-task deep neural networks outperformed classical single-task methods, offering the potential for conducting large-scale virtual screening, predicting activity profiles, and bridging the gaps in the available data.
深度学习是一种机器学习技术,通过利用由多个经历各种线性和非线性变换的处理层组成的图,对数据中的高级抽象进行建模。已证明该技术在药物发现应用中表现良好,可利用小分子的结构特征来预测活性。在此,我们报告一项大规模研究,以预测小分子在人类激酶组(一个主要的药物靶点家族,尤其是在抗癌药物中)中的活性。虽然小分子激酶抑制剂在几种不同疾病中展现出令人印象深刻的临床疗效,但耐药性通常会通过适应性激酶组重编程或亚群多样性而产生。多靶点药理学和联合疗法为耐药性疾病患者提供了潜在的治疗策略。它们的发展将受益于对人类激酶组中小分子抑制作用更全面和密集的了解。利用超过30万个小分子的65万多个生物活性注释,我们评估了多种机器学习方法,以预测人类激酶组中342种激酶的小分子抑制作用。我们的结果表明,多任务深度神经网络优于经典的单任务方法,为进行大规模虚拟筛选、预测活性谱以及弥补现有数据中的差距提供了潜力。