Epidemiology and Public Health Group, Department of Clinical and Biomedical Sciences, Faculty of Health and Life Sciences, University of Exeter, Exeter, UK.
Department of Gastroenterology, South Warwickshire University NHS Foundation Trust, Warwick, UK.
BMJ Open. 2024 Mar 13;14(3):e081926. doi: 10.1136/bmjopen-2023-081926.
haemochromatosis genetic variants have an uncertain clinical penetrance, especially to older ages and in undiagnosed groups. We estimated p.C282Y and p.H63D variant cumulative incidence of multiple clinical outcomes in a large community cohort.
Prospective cohort study.
22 assessment centres across England, Scotland, and Wales in the UK Biobank (2006-2010).
451 270 participants genetically similar to the 1000 Genomes European reference population, with a mean of 13.3-year follow-up through hospital inpatient, cancer registries and death certificate data.
Cox proportional HRs of incident clinical outcomes and mortality in those with p.C282Y/p.H63D mutations compared with those with no variants, stratified by sex and adjusted for age, assessment centre and genetic stratification. Cumulative incidences were estimated from age 40 years to 80 years.
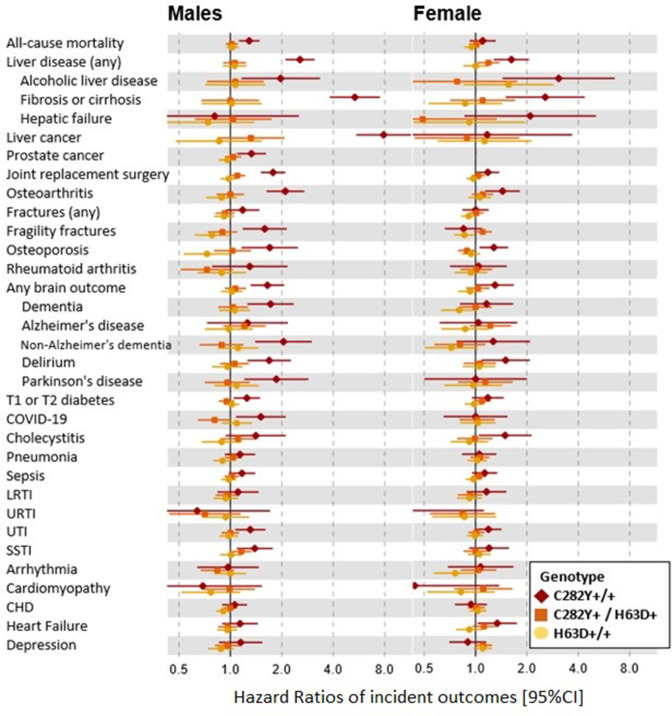
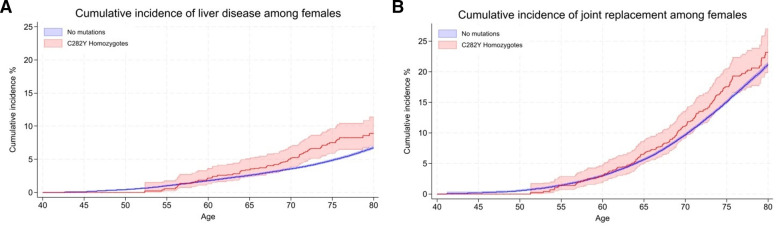
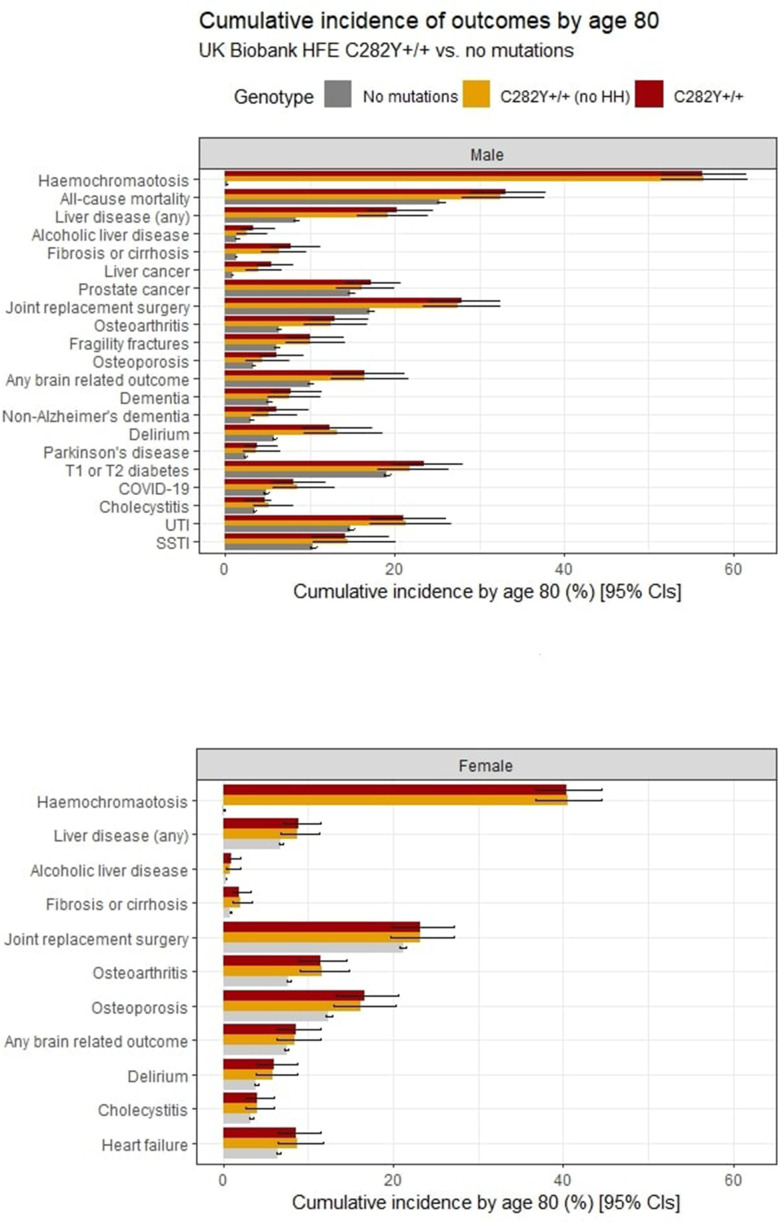
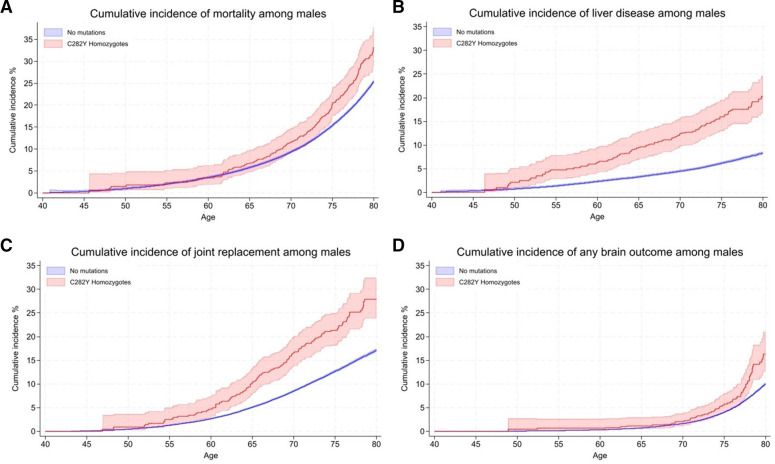
12.1% of p.C282Y+/+ males had baseline (mean age 57 years) haemochromatosis diagnoses, with a cumulative incidence of 56.4% at age 80 years. 33.1% died vs 25.4% without variants (HR 1.29, 95% CI: 1.12 to 1.48, p=4.7×10); 27.9% vs 17.1% had joint replacements, 20.3% vs 8.3% had liver disease, and there were excess delirium, dementia, and Parkinson's disease but not depression. Associations, including excess mortality, were similar in the group undiagnosed with haemochromatosis. 3.4% of women with p.C282Y+/+ had baseline haemochromatosis diagnoses, with a cumulative incidence of 40.5% at age 80 years. There were excess incident liver disease (8.9% vs 6.8%; HR 1.62, 95% CI: 1.27 to 2.05, p=7.8×10), joint replacements and delirium, with similar results in the undiagnosed. p.C282Y/p.H63D and p.H63D+/+ men or women had no statistically significant excess fatigue or depression at baseline and no excess incident outcomes.
Male and female p.C282Y homozygotes experienced greater excess morbidity than previously documented, including those undiagnosed with haemochromatosis in the community. As haemochromatosis diagnosis rates were low at baseline despite treatment being considered effective, trials of screening to identify people with p.C282Y homozygosity early appear justified.
血色病基因突变的临床外显率不确定,尤其是在年龄较大和未确诊的人群中。我们在一个大型社区队列中估计了 p.C282Y 和 p.H63D 变异的多种临床结局的累积发生率。
前瞻性队列研究。
英国生物库中的 22 个评估中心分布在英格兰、苏格兰和威尔士(2006-2010 年)。
451270 名在遗传上与 1000 基因组欧洲参考人群相似的参与者,平均随访时间为 13.3 年,通过住院患者、癌症登记处和死亡证明数据进行随访。
与无变异者相比,p.C282Y/p.H63D 突变者发生临床结局和死亡的 Cox 比例风险比,按性别分层,并按年龄、评估中心和遗传分层进行调整。累积发生率从 40 岁到 80 岁进行估计。
12.1%的 p.C282Y+/+男性基线(平均年龄 57 岁)有血色病诊断,80 岁时累积发病率为 56.4%。33.1%死亡与无变异者的 25.4%相比(HR 1.29,95%CI:1.12 至 1.48,p=4.7×10);27.9%与 17.1%有关节置换,20.3%与 8.3%有肝病,并有更多的谵妄、痴呆和帕金森病,但没有抑郁症。包括过度死亡在内的关联在未确诊为血色病的患者中相似。3.4%的 p.C282Y+/+女性基线时有血色病诊断,80 岁时累积发病率为 40.5%。肝病(8.9% vs 6.8%;HR 1.62,95%CI:1.27 至 2.05,p=7.8×10)、关节置换和谵妄的发病率过高,未确诊患者也有类似结果。p.C282Y/p.H63D 和 p.H63D+/+男性或女性在基线时没有明显的疲劳或抑郁过度,也没有更多的新发结局。
p.C282Y 纯合子男性和女性的发病率高于之前报道的发病率,包括社区中未确诊的血色病患者。尽管治疗被认为是有效的,但基线时的血色病诊断率较低,因此早期筛查识别 p.C282Y 纯合子的人群进行筛查的试验似乎是合理的。