Anderson Amelia, Piñeiro Ángel, García-Fandiño Rebeca, O'Connor Matthew S
Cyclarity Therapeutics, 8001 Redwood Blvd, Novato, CA 94945, USA.
Department of Organic Chemistry, Center for Research in Biological Chemistry and Molecular Materials, Santiago de Compostela University, CIQUS, Spain.
Comput Struct Biotechnol J. 2024 Feb 16;23:1117-1128. doi: 10.1016/j.csbj.2024.02.011. eCollection 2024 Dec.
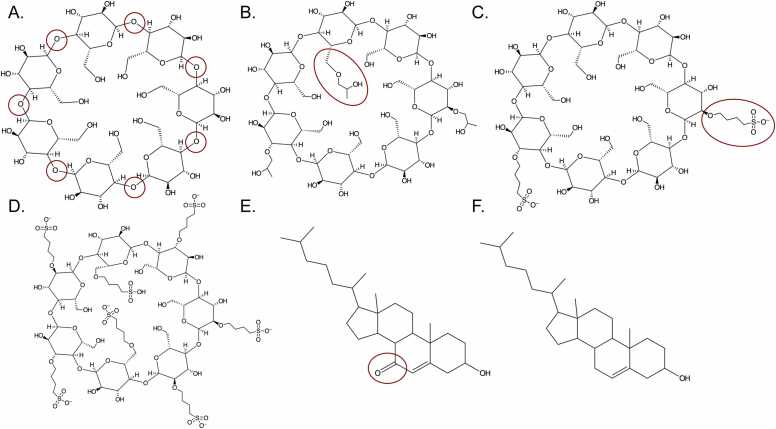
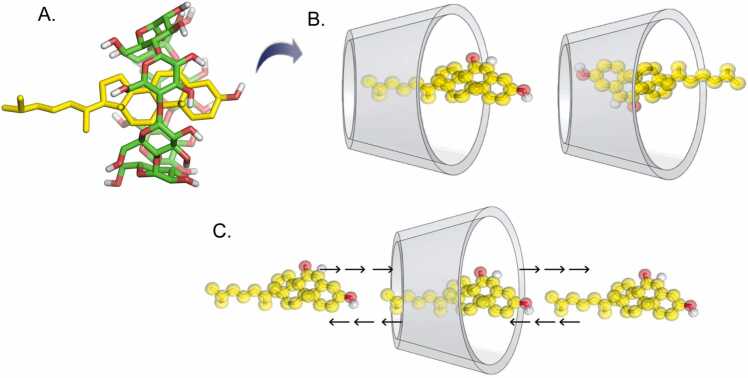
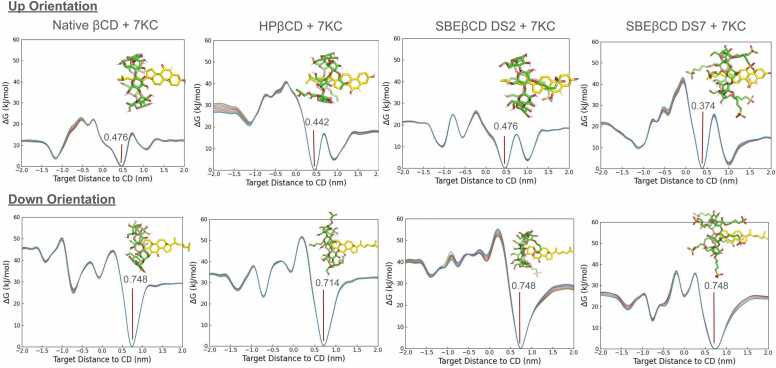
Cyclodextrins (CDs) are cyclic carbohydrate polymers that hold significant promise for drug delivery and industrial applications. Their effectiveness depends on their ability to encapsulate target molecules with strong affinity and specificity, but quantifying affinities in these systems accurately is challenging for a variety of reasons. Computational methods represent an exceptional complement to assays because they can be employed for existing and hypothetical molecules, providing high resolution structures in addition to a mechanistic, dynamic, kinetic, and thermodynamic characterization. Here, we employ potential of mean force (PMF) calculations obtained from guided metadynamics simulations to characterize the 1:1 inclusion complexes between four different modified βCDs, with different type, number, and location of substitutions, and two sterol molecules (cholesterol and 7-ketocholesterol). Our methods, validated for reproducibility through four independent repeated simulations per system and different post processing techniques, offer new insights into the formation and stability of CD-sterol inclusion complexes. A systematic distinct orientation preference where the sterol tail projects from the CD's larger face and significant impacts of CD substitutions on binding are observed. Notably, sampling only the CD cavity's wide face during simulations yielded comparable binding energies to full-cavity sampling, but in less time and with reduced statistical uncertainty, suggesting a more efficient approach. Bridging computational methods with complex molecular interactions, our research enables predictive CD designs for diverse applications. Moreover, the high reproducibility, sensitivity, and cost-effectiveness of the studied methods pave the way for extensive studies of massive CD-ligand combinations, enabling AI algorithm training and automated molecular design.
环糊精(CDs)是环状碳水化合物聚合物,在药物递送和工业应用方面具有巨大潜力。它们的有效性取决于其以强亲和力和特异性包封靶分子的能力,但由于多种原因,准确量化这些系统中的亲和力具有挑战性。计算方法是实验的出色补充,因为它们可用于现有分子和假设分子,除了提供机理、动力学、动态和热力学表征外,还能提供高分辨率结构。在这里,我们利用从引导式元动力学模拟获得的平均力势(PMF)计算来表征四种不同修饰的β-环糊精(具有不同类型、数量和取代位置)与两种甾醇分子(胆固醇和7-酮胆固醇)之间的1:1包合物。我们的方法通过对每个系统进行四次独立重复模拟和不同的后处理技术验证了其可重复性,为CD-甾醇包合物的形成和稳定性提供了新的见解。观察到一种系统的明显取向偏好,即甾醇尾部从CD的较大面伸出,以及CD取代对结合的显著影响。值得注意的是,在模拟过程中仅对CD腔的宽面进行采样产生的结合能与全腔采样相当,但所需时间更短且统计不确定性更低,这表明这是一种更有效的方法。我们的研究将计算方法与复杂的分子相互作用相结合,实现了针对各种应用的预测性CD设计。此外,所研究方法的高可重复性、灵敏度和成本效益为大规模CD-配体组合的广泛研究铺平了道路,能够进行人工智能算法训练和自动化分子设计。