Department of Chemical Engineering, Istanbul Technical University, Istanbul 34469, Turkey.
Institute of Human Virology, University of Maryland School of Medicine, Baltimore, Maryland 21201, United States.
J Phys Chem B. 2024 May 30;128(21):5157-5174. doi: 10.1021/acs.jpcb.4c00925. Epub 2024 Apr 22.
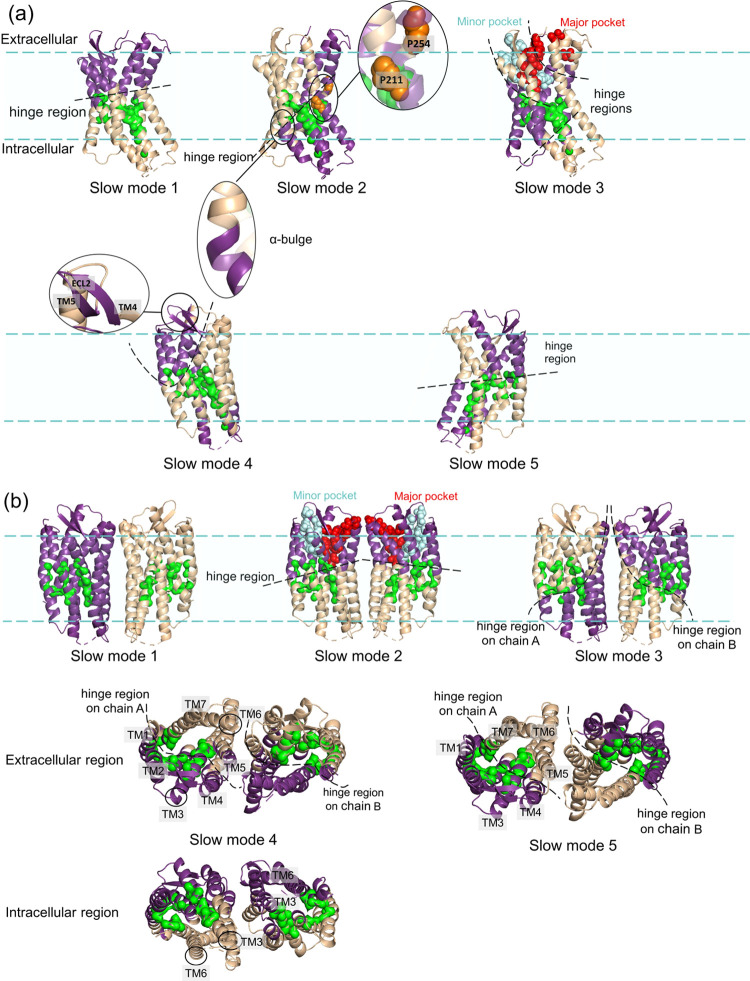
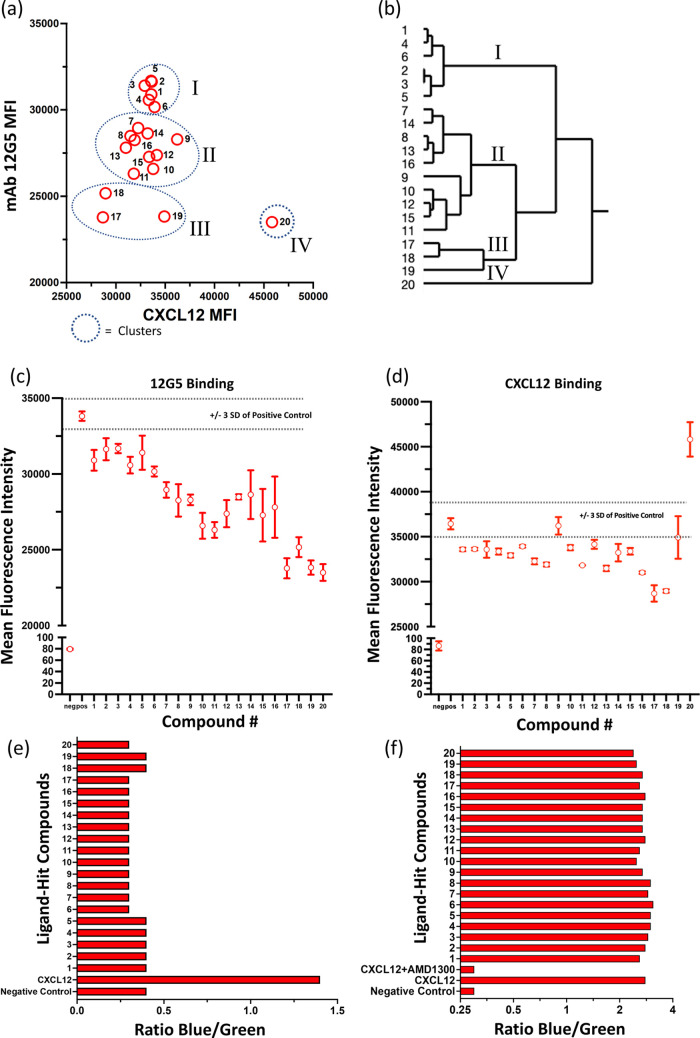
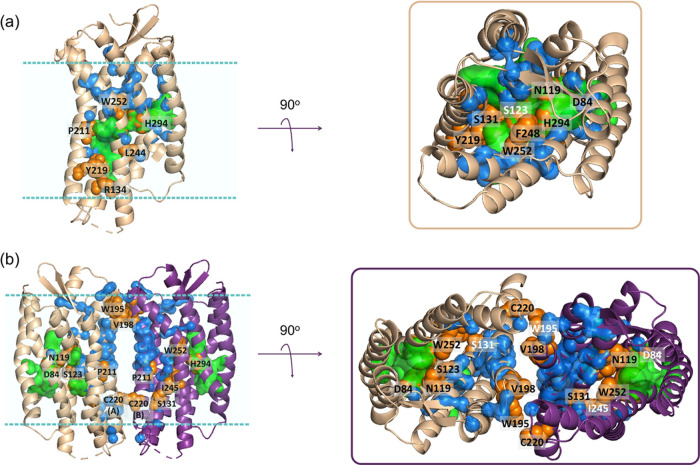
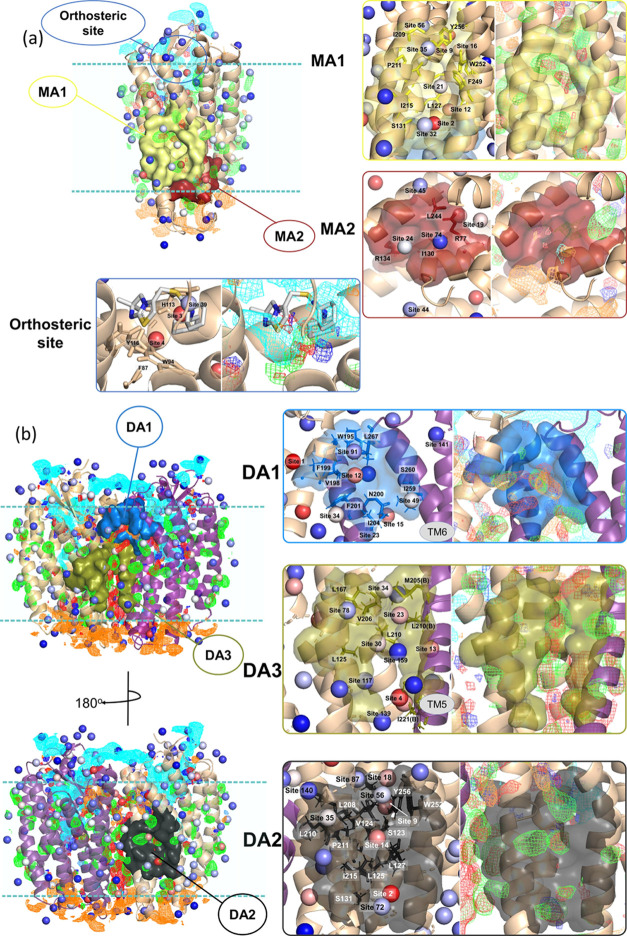
The chemokine receptor CXCR4 is a critical target for the treatment of several cancer types and HIV-1 infections. While orthosteric and allosteric modulators have been developed targeting its extracellular or transmembrane regions, the intramembrane region of CXCR4 may also include allosteric binding sites suitable for the development of allosteric drugs. To investigate this, we apply the Gaussian Network Model (GNM) to the monomeric and dimeric forms of CXCR4 to identify residues essential for its local and global motions located in the hinge regions of the protein. Residue interaction network (RIN) analysis suggests hub residues that participate in allosteric communication throughout the receptor. Mutual residues from the network models reside in regions with a high capacity to alter receptor dynamics upon ligand binding. We then investigate the druggability of these potential allosteric regions using the site identification by ligand competitive saturation (SILCS) approach, revealing two putative allosteric sites on the monomer and three on the homodimer. Two screening campaigns with Glide and SILCS-Monte Carlo docking using FDA-approved drugs suggest 20 putative hit compounds including antifungal drugs, anticancer agents, HIV protease inhibitors, and antimalarial drugs. assays considering mAB 12G5 and CXCL12 demonstrate both positive and negative allosteric activities of these compounds, supporting our computational approach. However, functional assays based on the recruitment of β-arrestin to CXCR4 do not show significant agonism and antagonism at a single compound concentration. The present computational pipeline brings a new perspective to computer-aided drug design by combining conformational dynamics based on network analysis and cosolvent analysis based on the SILCS technology to identify putative allosteric binding sites using CXCR4 as a showcase.
趋化因子受体 CXCR4 是治疗多种癌症类型和 HIV-1 感染的关键靶点。虽然已经开发出针对其细胞外或跨膜区域的变构调节剂和正构调节剂,但 CXCR4 的跨膜区域内也可能包括适合开发变构药物的变构结合位点。为了研究这一点,我们将高斯网络模型 (GNM) 应用于 CXCR4 的单体和二聚体形式,以确定位于蛋白质铰链区域的对其局部和全局运动至关重要的残基。残基相互作用网络 (RIN) 分析表明,参与整个受体变构通讯的枢纽残基。网络模型的相互残基位于配体结合后改变受体动力学能力较高的区域。然后,我们使用配体竞争饱和的位点鉴定 (SILCS) 方法研究这些潜在变构区域的可成药性,揭示单体上的两个假定变构位点和同源二聚体上的三个。使用 Glide 和 SILCS-Monte Carlo 对接进行的两次筛选实验,使用 FDA 批准的药物,共提出了 20 种潜在的命中化合物,包括抗真菌药物、抗癌药物、HIV 蛋白酶抑制剂和抗疟疾药物。考虑到 mAB 12G5 和 CXCL12 的 实验表明,这些化合物具有正变构和负变构活性,支持我们的计算方法。然而,基于 CXCR4 募集β-arrestin 的功能实验并未显示单一化合物浓度下的显著激动和拮抗作用。本计算流水线通过结合基于网络分析的构象动力学和基于 SILCS 技术的溶剂化分析,为计算机辅助药物设计带来了新的视角,使用 CXCR4 作为范例来识别潜在的变构结合位点。