Peng Jie, Gu Yawen, Liu Jiang, Yi Hao, Ruan Dong, Huang Haoyu, Shu Yuan, Zong Zhen, Wu Rui, Li Hui
Department of Rheumatology and Immunology, the First Affiliated Hospital, Jiangxi Medical College, Nanchang University, 330006, Nanchang, China.
The Second Clinical Medical College, Jiangxi Medical College, Nanchang University, 330006, Nanchang, China.
Heliyon. 2024 Apr 23;10(9):e30020. doi: 10.1016/j.heliyon.2024.e30020. eCollection 2024 May 15.
Gout is the most common inflammatory arthritis in adults. Gout is an arthritic disease caused by the deposition of monosodium urate crystal (MSU) in the joints, which can lead to acute inflammation and damage adjacent tissue. Hyperuricemia is the main risk factor for MSU crystal deposition and gout. With the increasing burden of gout disease, the identification of potential biomarkers and novel targets for diagnosis is urgently needed.
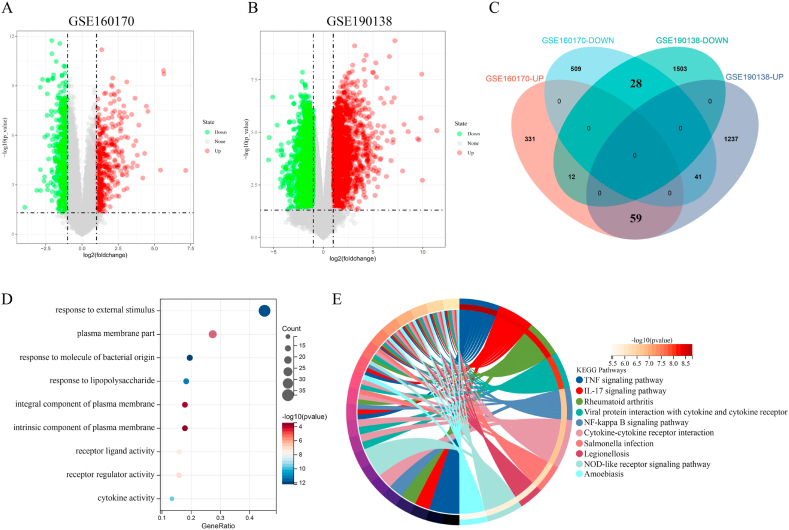
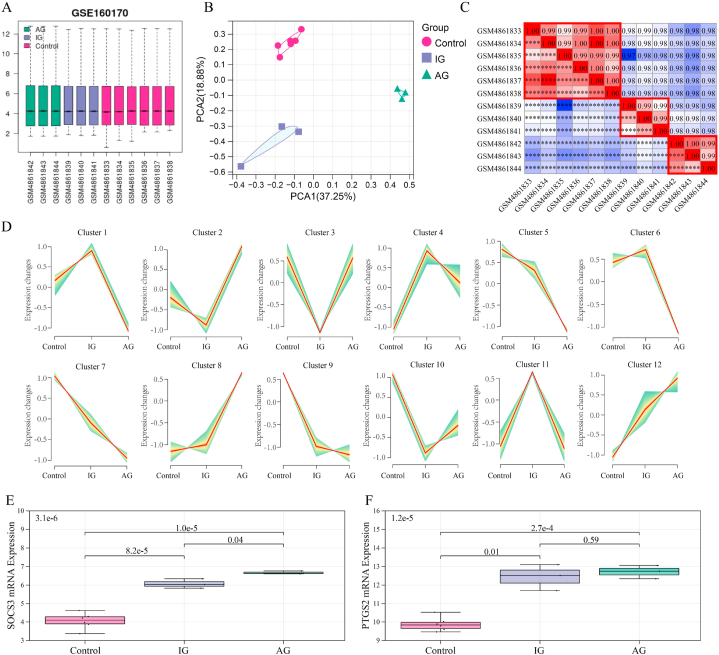
For the analysis of this subject paper, we downloaded the human gout data set GSE160170 and the gout mouse model data set GSE190138 from the GEO database. To obtain the differentially expressed genes (DEGs), we intersected the two data sets. Using the cytohubba algorithm, we identified the key genes and enriched them through GO and KEGG. The gene expression trends of three subgroups (normal control group, intermittent gout group and acute gout attack group) were analyzed by Series Test of Cluster (STC) analysis, and the key genes were screened out, and the diagnostic effect was verified by ROC curve. The expression of key genes in dorsal root nerve and spinal cord of gout mice was analyzed. Finally, the clinical samples of normal control group, hyperuricemia group, intermittent gout group and acute gout attack group were collected, and the expression of key genes at protein level was verified by ELISA.
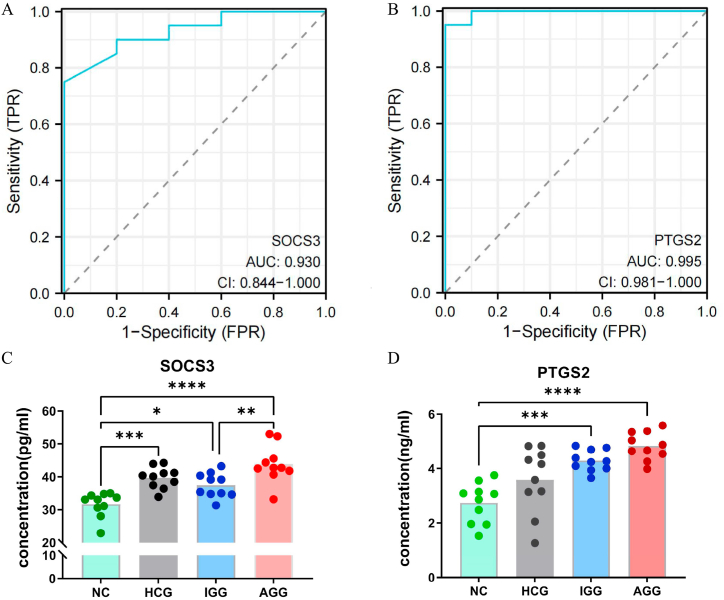
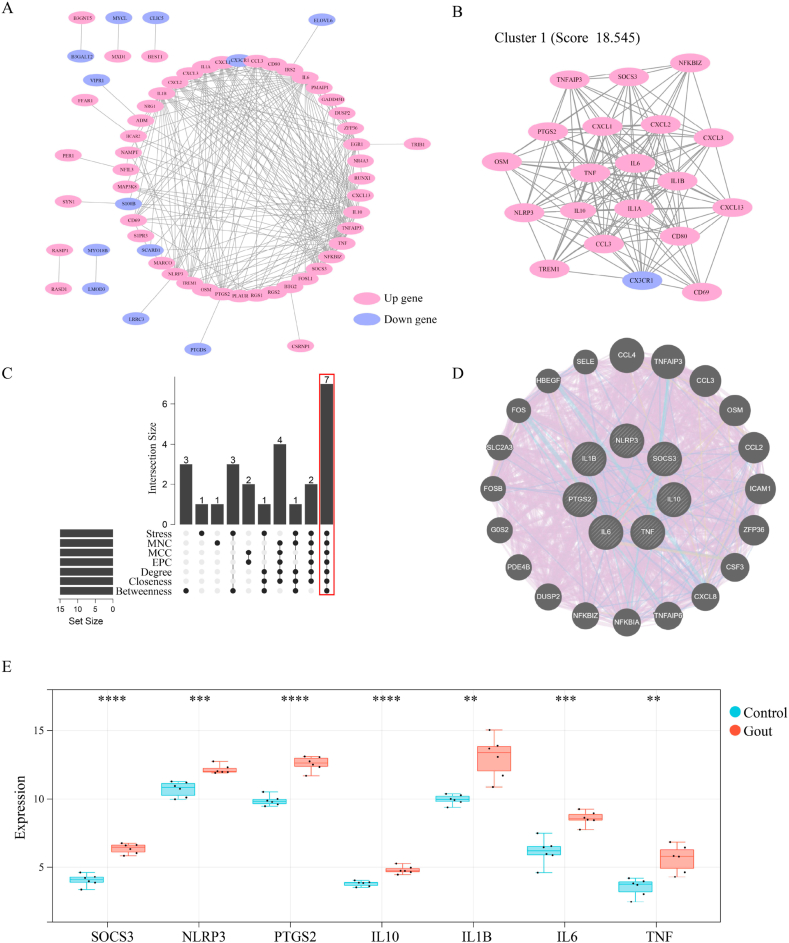
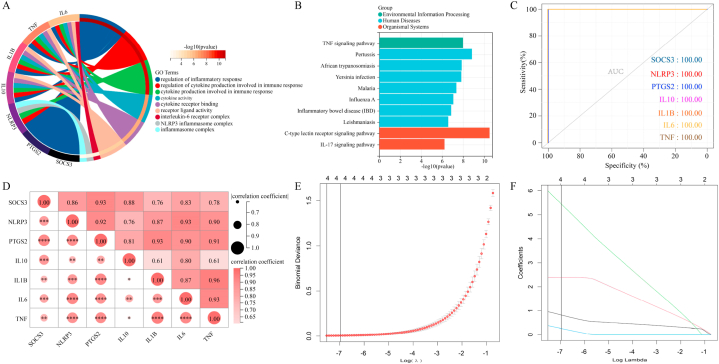
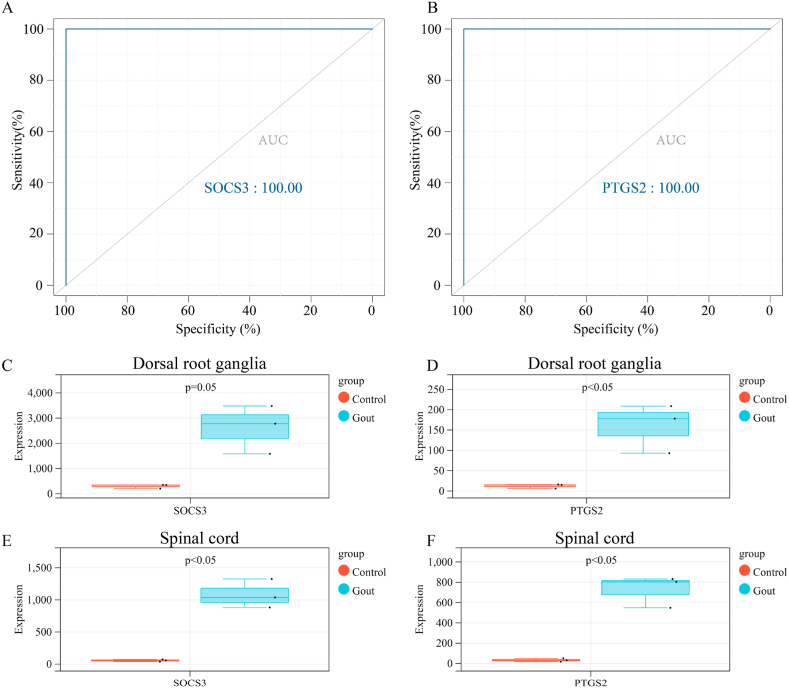
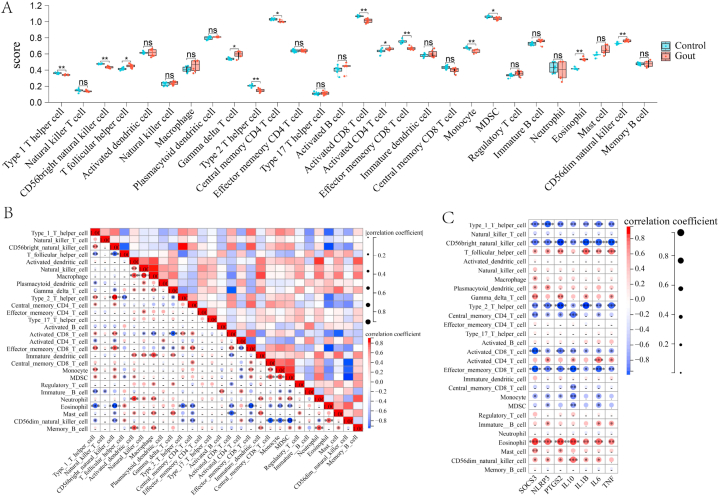
We obtained 59 co-upregulated and 28 co-downregulated genes by comparing the DEGs between gout mouse model data set and human gout data set. 7 hub DEGs(IL1B, IL10, NLRP3, SOCS3, PTGS2) were screened out via Cytohubba algorithm. The results of both GO and KEGG enrichment analyses indicate that 7 hub genes play a significant role in regulating the inflammatory response, cytokine production in immune response, and the TNF signaling pathway. The most representative hub genes SOCS3 and PTGS2 were screened out by Series Test of Cluster, and ROC analysis results showed the AUC values were both up to 1.000. In addition, we found that PTGS2 expression was significantly elevated in the dorsal root ganglia and spinal cord in monosodium urate(MSU)-induced gout mouse model. The ELISA results revealed that the expression of SOCS3 and PTGS2 was notably higher in the acute gout attack and intermittent gout groups compared to the normal control group. This difference was statistically significant, indicating a clear distinction between the groups.
Through cross-species comprehensive analysis and experimental verification, SOCS3 and PTGS2 were proved to be new biomarkers for diagnosing gout and predicting disease progression.
痛风是成年人中最常见的炎性关节炎。痛风是一种由尿酸单钠晶体(MSU)在关节中沉积引起的关节炎疾病,可导致急性炎症并损害邻近组织。高尿酸血症是MSU晶体沉积和痛风的主要危险因素。随着痛风疾病负担的增加,迫切需要鉴定潜在的生物标志物和新的诊断靶点。
为了分析本主题论文,我们从GEO数据库下载了人类痛风数据集GSE160170和痛风小鼠模型数据集GSE190138。为了获得差异表达基因(DEG),我们将这两个数据集进行了交叉分析。使用Cytohubba算法,我们鉴定了关键基因,并通过GO和KEGG对其进行了富集分析。通过聚类序列检验(STC)分析了三个亚组(正常对照组、间歇性痛风组和急性痛风发作组)的基因表达趋势,筛选出关键基因,并通过ROC曲线验证了诊断效果。分析了痛风小鼠背根神经和脊髓中关键基因的表达。最后,收集了正常对照组、高尿酸血症组、间歇性痛风组和急性痛风发作组的临床样本,并通过ELISA验证了关键基因在蛋白水平的表达。
通过比较痛风小鼠模型数据集和人类痛风数据集之间的DEG,我们获得了59个共上调基因和28个共下调基因。通过Cytohubba算法筛选出7个核心DEG(IL1B、IL10、NLRP3、SOCS3、PTGS2)。GO和KEGG富集分析结果均表明,7个核心基因在调节炎症反应、免疫反应中的细胞因子产生以及TNF信号通路中发挥着重要作用。通过聚类序列检验筛选出最具代表性的核心基因SOCS3和PTGS2,ROC分析结果显示AUC值均高达1.000。此外,我们发现尿酸单钠(MSU)诱导的痛风小鼠模型中,背根神经节和脊髓中PTGS2的表达显著升高。ELISA结果显示,与正常对照组相比,急性痛风发作组和间歇性痛风组中SOCS3和PTGS2的表达明显更高。这种差异具有统计学意义,表明各组之间存在明显区别。
通过跨物种综合分析和实验验证,证明SOCS3和PTGS2是诊断痛风和预测疾病进展的新生物标志物。