Institute of Biomedical Informatics, Graz University of Technology, Graz, Austria.
BMC Genomics. 2024 May 8;25(1):455. doi: 10.1186/s12864-024-10344-9.
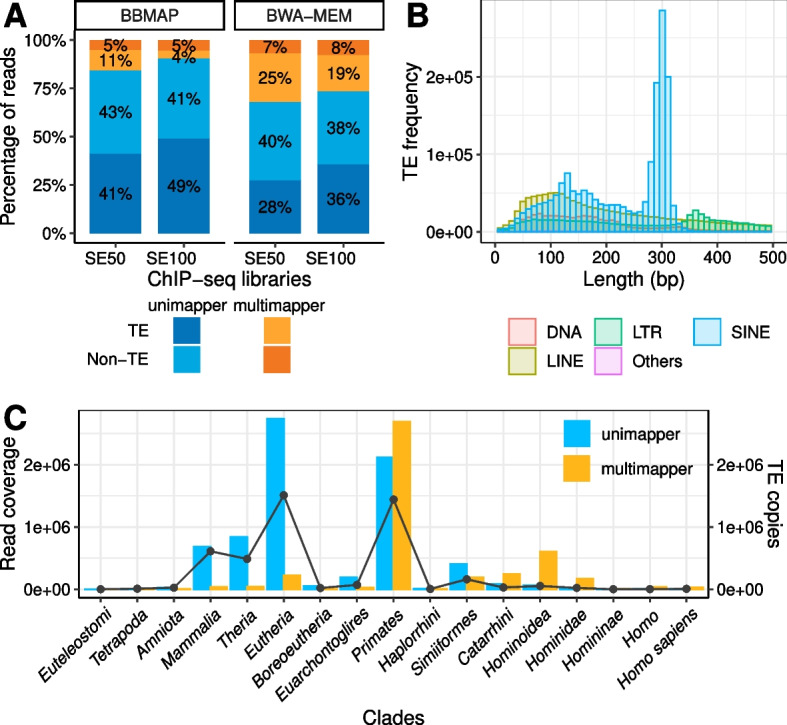
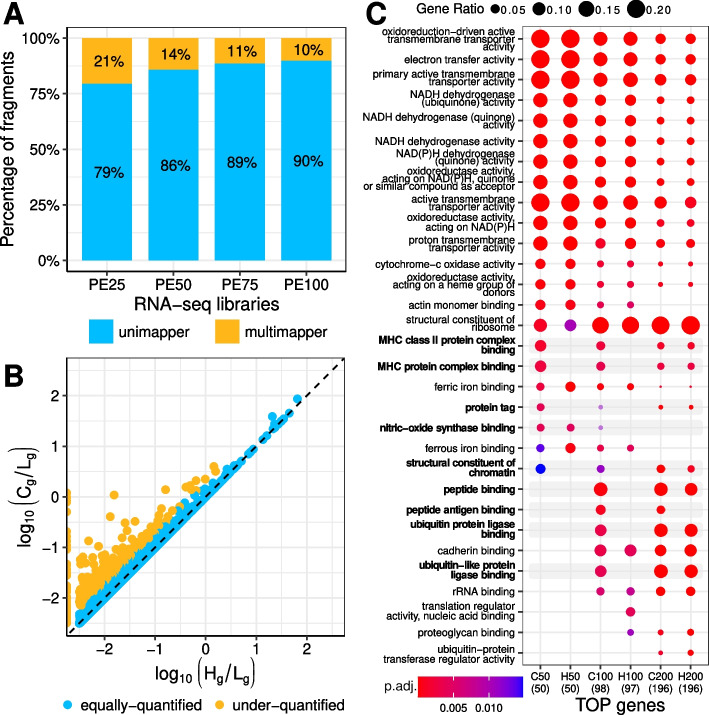
Standard ChIP-seq and RNA-seq processing pipelines typically disregard sequencing reads whose origin is ambiguous ("multimappers"). This usual practice has potentially important consequences for the functional interpretation of the data: genomic elements belonging to clusters composed of highly similar members are left unexplored.
In particular, disregarding multimappers leads to the underrepresentation in epigenetic studies of recently active transposable elements, such as AluYa5, L1HS and SVAs. Furthermore, this common strategy also has implications for transcriptomic analysis: members of repetitive gene families, such the ones including major histocompatibility complex (MHC) class I and II genes, are under-quantified.
Revealing inherent biases that permeate routine tasks such as functional enrichment analysis, our results underscore the urgency of broadly adopting multimapper-aware bioinformatic pipelines -currently restricted to specific contexts or communities- to ensure the reliability of genomic and transcriptomic studies.
标准 ChIP-seq 和 RNA-seq 处理管道通常会忽略其来源不明确的测序读取(“多重映射”)。这种常见的做法可能会对数据的功能解释产生重要影响:属于由高度相似成员组成的聚类的基因组元件未被探索。
特别是,忽略多重映射会导致在表观遗传学研究中,最近活跃的转座元件(如 AluYa5、L1HS 和 SVAs)的代表性不足。此外,这种常见策略也会对转录组分析产生影响:重复基因家族的成员,如主要组织相容性复合体(MHC)I 类和 II 类基因,被低估了。
揭示了普遍存在于功能富集分析等常规任务中的固有偏差,我们的结果强调了广泛采用多重映射感知生物信息学管道的紧迫性-目前仅限于特定的背景或社区-以确保基因组和转录组研究的可靠性。