Department of Cardiology Renmin Hospital of Wuhan University Wuhan China.
Hubei Key Laboratory of Metabolic and Chronic Diseases Wuhan China.
J Am Heart Assoc. 2024 May 21;13(10):e028006. doi: 10.1161/JAHA.122.028006. Epub 2024 May 10.
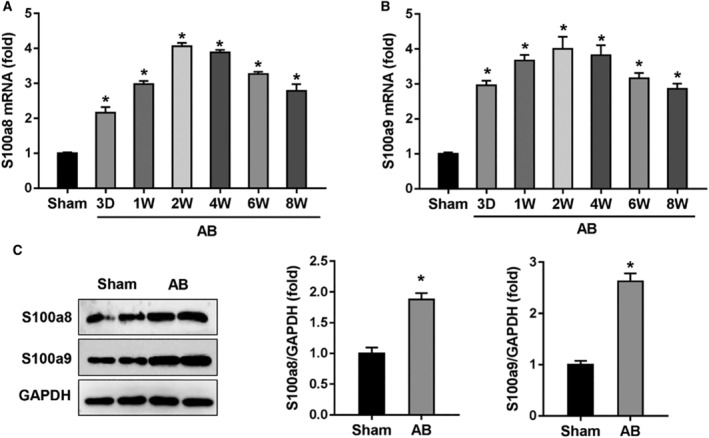
S100a8/9 (S100 calcium binding protein a8/9) belongs to the S100 family and has gained a lot of interest as a critical regulator of inflammatory response. Our previous study found that S100a8/9 homolog promoted aortic valve sclerosis in mice with chronic kidney disease. However, the role of S100a8/9 in pressure overload-induced cardiac hypertrophy remains unclear. The present study was to explore the role of S100a8/9 in cardiac hypertrophy.
Cardiomyocyte-specific S100a9 loss or gain of function was achieved using an adeno-associated virus system, and the model of cardiac hypertrophy was established by aortic banding-induced pressure overload. The results indicate that S100a8/9 expression was increased in response to pressure overload. S100a9 deficiency alleviated pressure overload-induced hypertrophic response, whereas S100a9 overexpression accelerated cardiac hypertrophy. S100a9-overexpressed mice showed increased FGF23 (fibroblast growth factor 23) expression in the hearts after exposure to pressure overload, which activated calcineurin/NFAT (nuclear factor of activated T cells) signaling in cardiac myocytes and thus promoted hypertrophic response. A specific antibody that blocks FGFR4 (FGF receptor 4) largely abolished the prohypertrophic response of S100a9 in mice.
In conclusion, S100a8/9 promoted the development of cardiac hypertrophy in mice. Targeting S100a8/9 may be a promising therapeutic approach to treat cardiac hypertrophy.
S100a8/9(S100 钙结合蛋白 a8/9)属于 S100 家族,作为炎症反应的关键调节因子备受关注。我们之前的研究发现,S100a8/9 同源物促进了慢性肾脏病小鼠的主动脉瓣硬化。然而,S100a8/9 在压力超负荷诱导的心肌肥厚中的作用尚不清楚。本研究旨在探讨 S100a8/9 在心肌肥厚中的作用。
利用腺相关病毒系统实现心肌细胞特异性 S100a9 缺失或功能获得,通过主动脉缩窄诱导压力超负荷建立心肌肥厚模型。结果表明,S100a8/9 的表达在压力超负荷时增加。S100a9 缺失减轻了压力超负荷诱导的心肌肥厚反应,而 S100a9 过表达加速了心肌肥厚。在暴露于压力超负荷后,S100a9 过表达的小鼠心脏中 FGF23(成纤维细胞生长因子 23)的表达增加,激活了心肌细胞中的钙调神经磷酸酶/NFAT(激活 T 细胞的核因子)信号通路,从而促进了心肌肥厚反应。一种特异性阻断 FGFR4(FGF 受体 4)的抗体在很大程度上消除了 S100a9 在小鼠中的促肥厚反应。
总之,S100a8/9 促进了小鼠心肌肥厚的发展。靶向 S100a8/9 可能是治疗心肌肥厚的一种有前途的治疗方法。