Chimatahalli Shanthakumar Karthik, Sridhara Pruthvishree Guluvinaattiguppe, Rajabathar Jothi Ramalingam, Al-Lohedan Hamad A, Lokanath Neratur Krishnappagowda, Mylnahalli Krishnegowda Hema
Department of Chemistry, SJCE, JSS Science and Technology University, Mysuru, Karnataka 570 006, India.
Department of Studies in Physics, University of Mysore, Manasagangotri, Mysuru 570 006, India.
ACS Omega. 2024 May 3;9(19):20753-20772. doi: 10.1021/acsomega.3c07520. eCollection 2024 May 14.
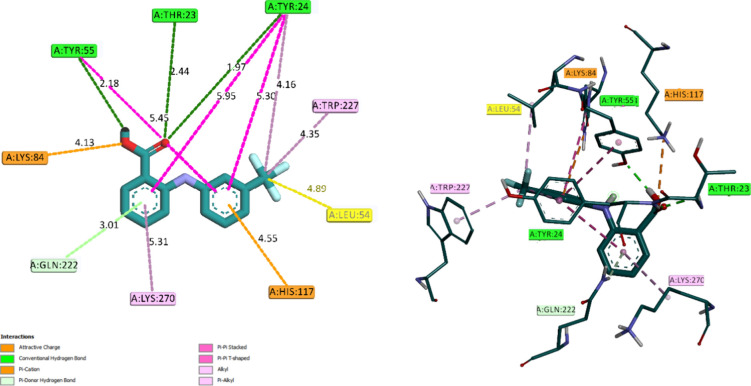
This paper delves into the polymorphism of 2-[3-(trifluoromethyl)anilino]benzoic acid, commonly referred to as flufenamic acid (FA), a pharmaceutical agent employed in treating inflammatory conditions. The central focus of the study is on a newly unearthed solvatomorphic structure of FA in methanol (FAM), and a thorough comparison is conducted with the commercially available standard structure. Employing a comprehensive approach, including X-ray crystallography, Hirshfeld surface analysis, density functional theory (DFT), molecular docking, and molecular dynamics (MD) simulations, the research aims to unravel the structural and functional implications of solvatomorphism. The X-ray crystal structure analysis brings to light notable differences between the standard FA and solvatomorphic FAM, showcasing variations in intermolecular interactions and crystal packing. Key features such as hydrogen bonding, π···π stacking, and C-H···π interactions are identified as influential factors shaping the stability and conformation of the compounds. Hirshfeld surface analysis further quantifies the nature and contribution of intermolecular interactions, providing a comprehensive perspective on molecular stability. Density functional theory offers valuable electronic structure insights, highlighting disparities in frontier molecular orbitals between FA and FAM. Molecular docking studies against prostaglandin D2 11-ketoreductase explore potential drug interactions, unveiling distinct binding modes and hydrogen bonding patterns that shed light on how the solvatomorphic structure may impact drug-target interactions. In-depth molecular dynamics simulations over 100 ns investigate the stability of the protein-ligand complex, with root mean square deviation and root mean square fluctuation analyses revealing minimal deviations and affirming the stability of FAM within the active site of the target protein.
本文深入研究了2-[3-(三氟甲基)苯胺基]苯甲酸(通常称为氟芬那酸,简称FA)的多晶型现象,氟芬那酸是一种用于治疗炎症性疾病的药物。该研究的核心聚焦于新发现的氟芬那酸在甲醇中的溶剂同形结构(FAM),并与市售标准结构进行了全面比较。研究采用了包括X射线晶体学、 Hirshfeld表面分析、密度泛函理论(DFT)、分子对接和分子动力学(MD)模拟在内的综合方法,旨在揭示溶剂同形现象的结构和功能影响。X射线晶体结构分析揭示了标准FA和溶剂同形FAM之间的显著差异,展示了分子间相互作用和晶体堆积的变化。氢键、π···π堆积和C-H···π相互作用等关键特征被确定为影响化合物稳定性和构象的因素。Hirshfeld表面分析进一步量化了分子间相互作用的性质和贡献,提供了关于分子稳定性的全面视角。密度泛函理论提供了有价值的电子结构见解,突出了FA和FAM之间前沿分子轨道的差异。针对前列腺素D2 11-酮还原酶的分子对接研究探索了潜在的药物相互作用,揭示了不同的结合模式和氢键模式,从而阐明了溶剂同形结构可能如何影响药物-靶点相互作用。超过100纳秒的深入分子动力学模拟研究了蛋白质-配体复合物的稳定性,均方根偏差和均方根波动分析显示偏差极小,证实了FAM在靶蛋白活性位点内的稳定性。