Gorostidi-Aicua Miriam, Reparaz Iraia, Otaegui-Chivite Ane, García Koldo, Romarate Leire, Álvarez de Arcaya Amaya, Mendiburu Idoia, Arruti Maialen, Castillo-Triviño Tamara, Moles Laura, Otaegui David
Biogipuzkoa Health Research Institute, Neuroimmunology Group, 20014 San Sebastián, Spain.
Center for Biomedical Research Network in Neurodegenerative Diseases (CIBER-CIBERNED-ISCIII), 28029 Madrid, Spain.
Microorganisms. 2024 Apr 26;12(5):872. doi: 10.3390/microorganisms12050872.
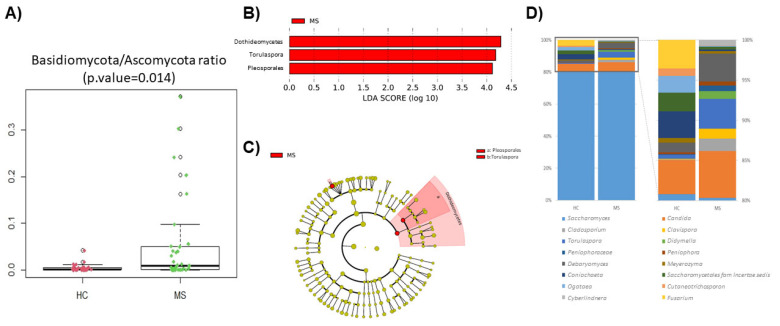
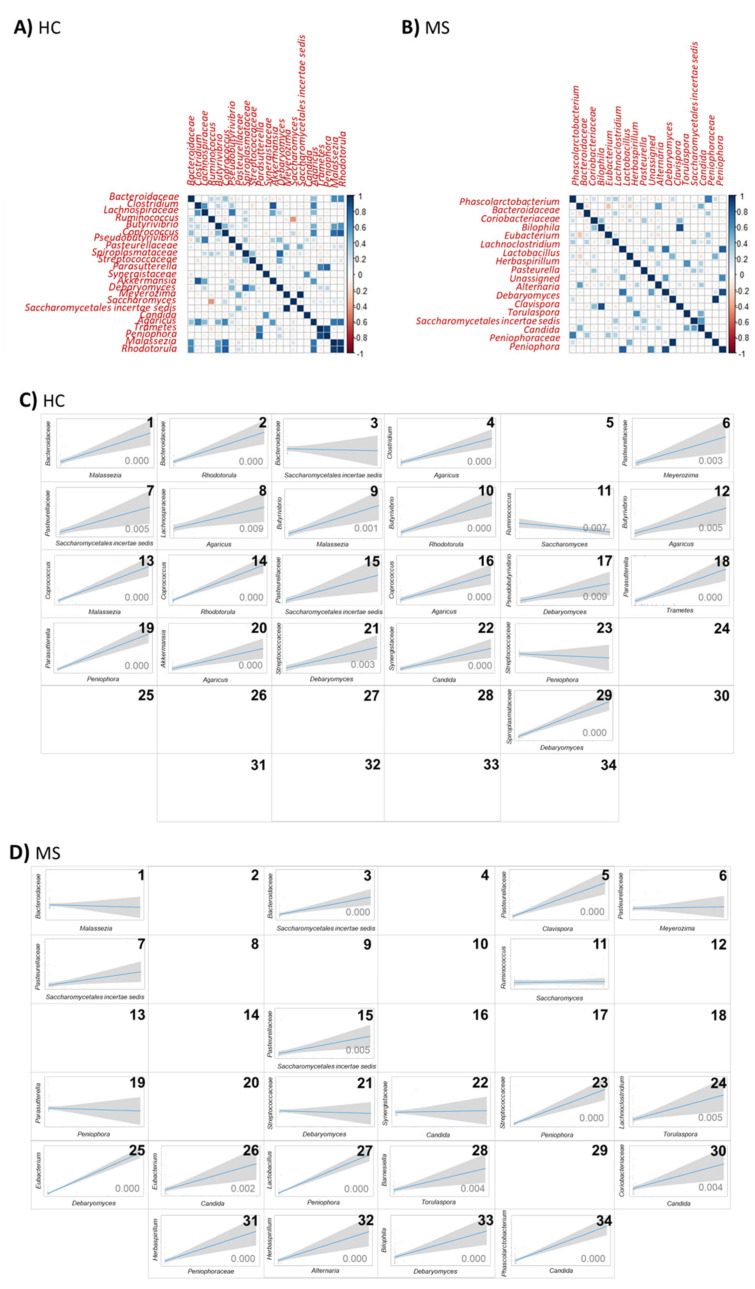
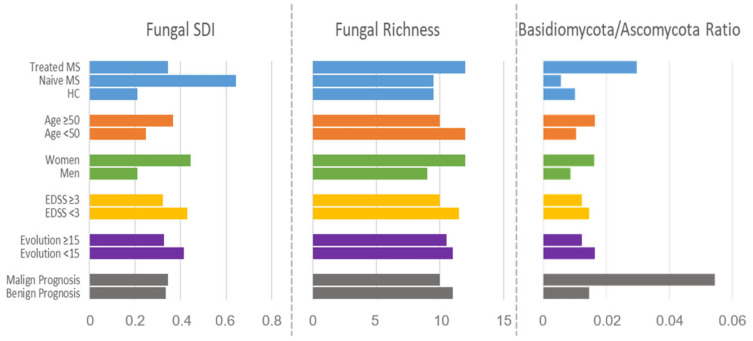
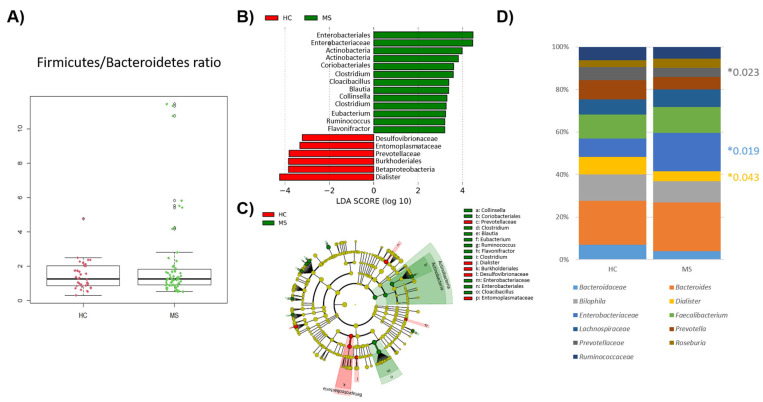
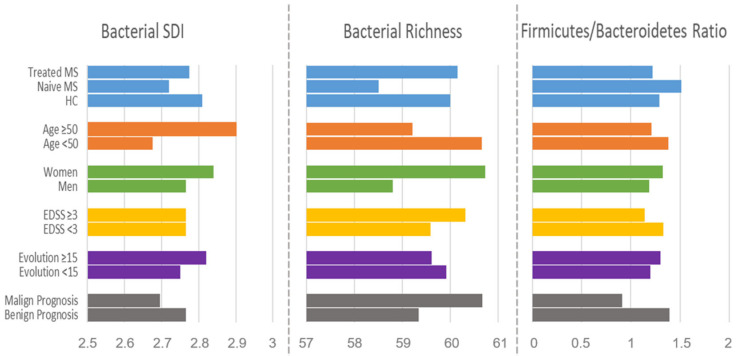
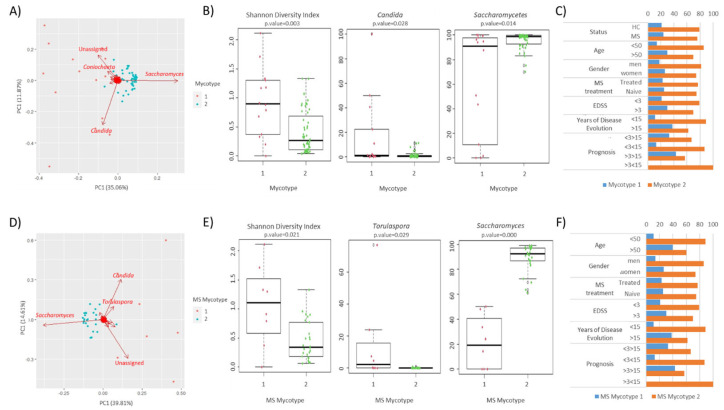
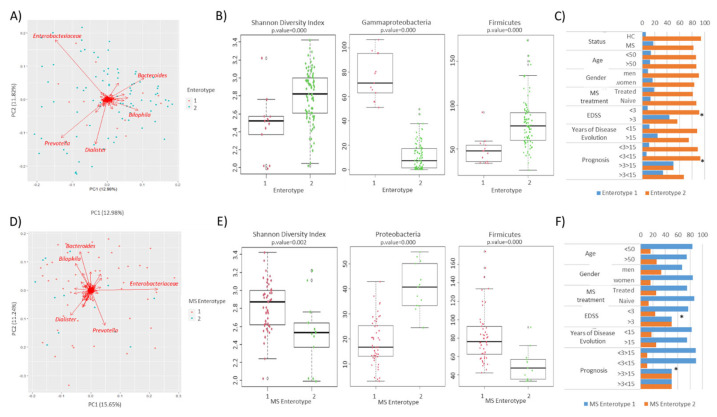
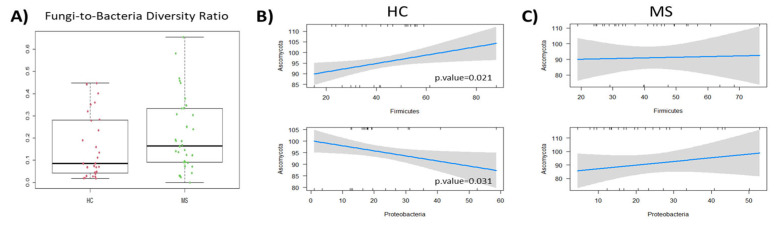
Multiple sclerosis (MS) arises from a complex interplay between host genetic factors and environmental components, with the gut microbiota emerging as a key area of investigation. In the current study, we used ion torrent sequencing to delve into the bacteriome (bacterial microbiota) and mycobiome (fungal microbiota) of people with MS (pwMS), and compared them to healthy controls (HC). Through principal coordinate, diversity, and abundance analyses, as well as clustering and cross-kingdom microbial correlation assessments, we uncovered significant differences in the microbial profiles between pwMS and HC. Elevated levels of the fungus and the bacterial family were observed in pwMS, whereas beneficial bacterial taxa, such as and , were reduced. Notably, clustering analysis revealed overlapping patterns in the bacteriome and mycobiome data for 74% of the participants, with weakened cross-kingdom interactions evident in the altered microbiota of pwMS. Our findings highlight the dysbiosis of both bacterial and fungal microbiota in MS, characterized by shifts in biodiversity and composition. Furthermore, the distinct disease-associated pattern of fungi-bacteria interactions suggests that fungi, in addition to bacteria, contribute to the pathogenesis of MS. Overall, our study sheds light on the intricate microbial dynamics underlying MS, paving the way for further investigation into the potential therapeutic targeting of the gut microbiota in MS management.
多发性硬化症(MS)源于宿主遗传因素与环境成分之间的复杂相互作用,肠道微生物群已成为一个关键的研究领域。在本研究中,我们使用离子激流测序技术深入研究了MS患者(pwMS)的细菌群落(细菌微生物群)和真菌群落(真菌微生物群),并将其与健康对照(HC)进行比较。通过主坐标分析、多样性分析和丰度分析,以及聚类分析和跨界微生物相关性评估,我们发现pwMS和HC之间的微生物谱存在显著差异。在pwMS中观察到真菌和细菌科的水平升高,而有益细菌类群,如 和 ,则减少。值得注意的是,聚类分析显示74%的参与者的细菌群落和真菌群落数据存在重叠模式,在pwMS改变的微生物群中,跨界相互作用减弱。我们的研究结果突出了MS中细菌和真菌微生物群的失调,其特征是生物多样性和组成的变化。此外,真菌与细菌相互作用的独特疾病相关模式表明,除了细菌外,真菌也促成了MS的发病机制。总体而言,我们的研究揭示了MS潜在的复杂微生物动态,为进一步研究肠道微生物群在MS治疗中的潜在靶向作用铺平了道路。