Huang Ronglei, Ji Xue, Zhu Lingwei, Zhang Chengyang, Luo Tingting, Liang Bing, Jiang Bowen, Zhou Ang, Du Chongtao, Sun Yang
State Key Laboratory for Diagnosis and Treatment of Severe Zoonotic Infectious Diseases, Key Laboratory for Zoonosis Research of the Ministry of Education, Institute of Zoonosis, College of Veterinary Medicine, Jilin University, Changchun 130062, China.
Changchun Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Changchun 130122, China.
Microorganisms. 2024 May 13;12(5):978. doi: 10.3390/microorganisms12050978.
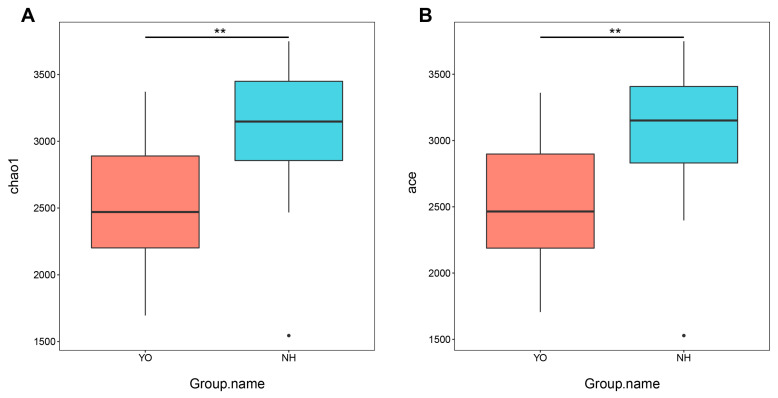
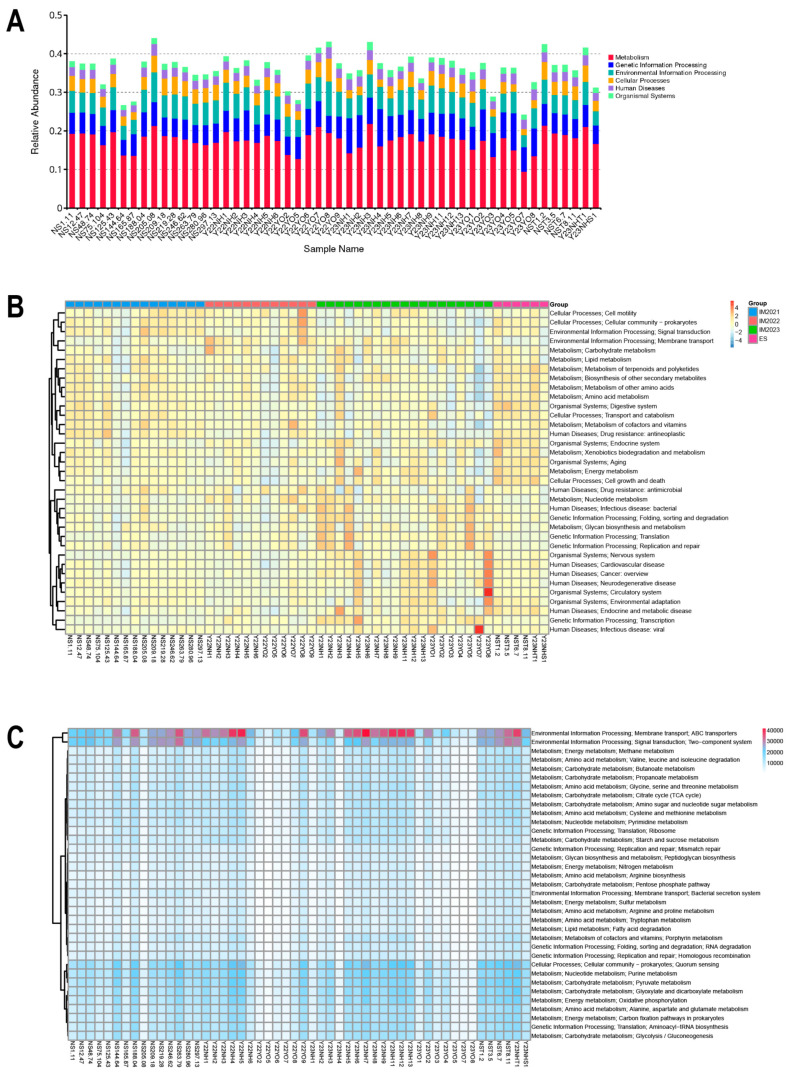
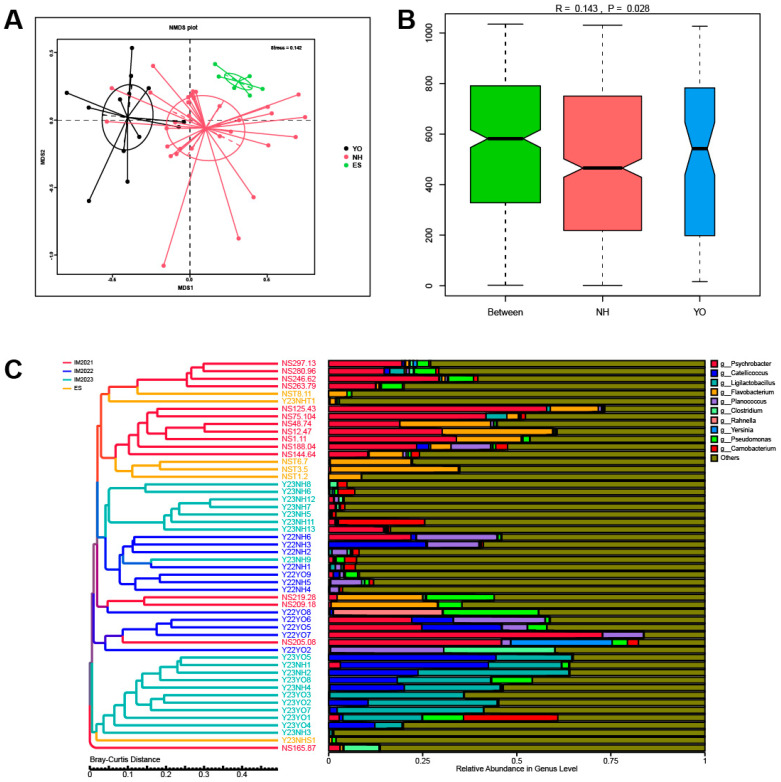
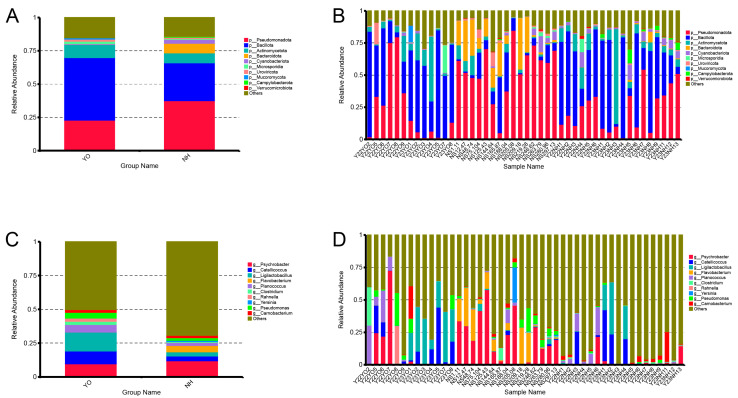
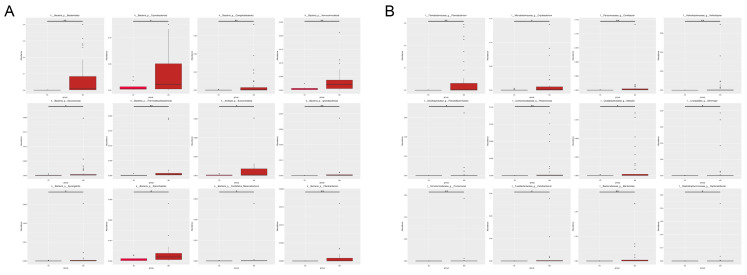
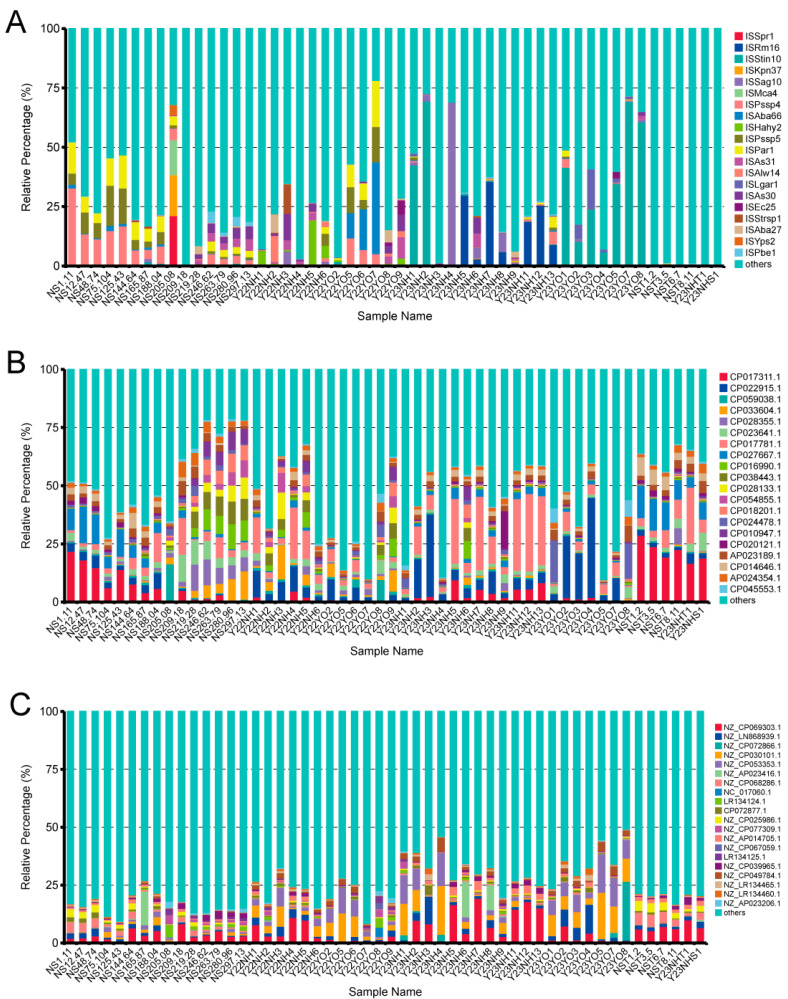
Gut microbes thrive by utilising host energy and, in return, provide valuable benefits, akin to a symbiotic relationship. Here, metagenomic sequencing was performed to characterise and compare the community composition, diversity and antibiotic resistance of the gut microbiota of Relict gull () and species. Alpha diversity analysis revealed that the intestinal microbial richness of was significantly lower than that of , with distinct differences observed in microbial composition. Notably, the intestines of harboured more pathogenic bacteria such as clostridium, which may contribute to the decline in their population and endangered status. A total of 117 strains of were isolated, with 90.60% exhibiting full susceptibility to 21 antibiotics, while 25.3% exhibited significant biofilm formation. Comprehensive Antibiotic Resistance Database data indicated that glycopeptide resistance genes were the most prevalent type carried by migratory birds, alongside quinolone, tetracycline and lincosamide resistance genes. The abundance of resistance genes carried by migratory birds decreased over time. This metagenomic analysis provides valuable insights into the intestinal microbial composition of these wild bird species, offering important guidance for their conservation efforts, particularly for , and contributing to our understanding of pathogen spread and antibiotic-resistant bacteria.
肠道微生物通过利用宿主能量而蓬勃发展,作为回报,它们提供了宝贵的益处,类似于一种共生关系。在此,进行了宏基因组测序,以表征和比较遗鸥()和物种的肠道微生物群的群落组成、多样性和抗生素抗性。α多样性分析表明,的肠道微生物丰富度显著低于,且在微生物组成上观察到明显差异。值得注意的是,的肠道中含有更多的致病菌,如梭菌,这可能导致其种群数量下降和濒危状态。共分离出117株,其中90.60%对21种抗生素表现出完全敏感性,而25.3%表现出显著的生物膜形成。综合抗生素抗性数据库数据表明,糖肽抗性基因是候鸟携带的最普遍类型,同时还有喹诺酮、四环素和林可酰胺抗性基因。候鸟携带的抗性基因丰度随时间下降。这项宏基因组分析为这些野生鸟类物种的肠道微生物组成提供了有价值的见解,为它们的保护工作,特别是为的保护工作提供了重要指导,并有助于我们理解病原体传播和抗生素抗性细菌。