Lapp Zena, Freedman Elizabeth, Huang Kathie, Markwalter Christine F, Obala Andrew A, Prudhomme-O'Meara Wendy, Taylor Steve M
Duke Global Health Institute, Duke University, Durham, North Carolina, United States of America.
Division of Infectious Diseases, School of Medicine, Duke University, Durham, North Carolina, United States of America.
PLOS Glob Public Health. 2024 May 30;4(5):e0002361. doi: 10.1371/journal.pgph.0002361. eCollection 2024.
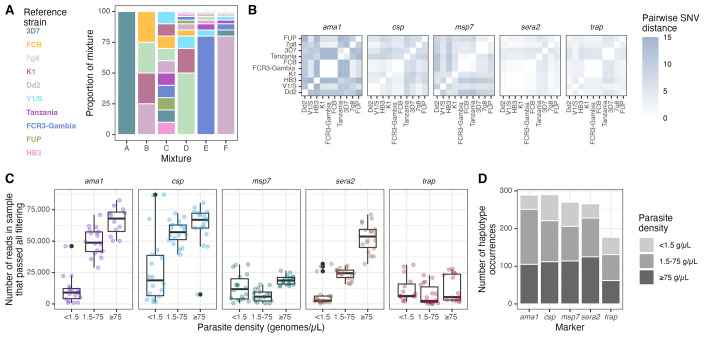
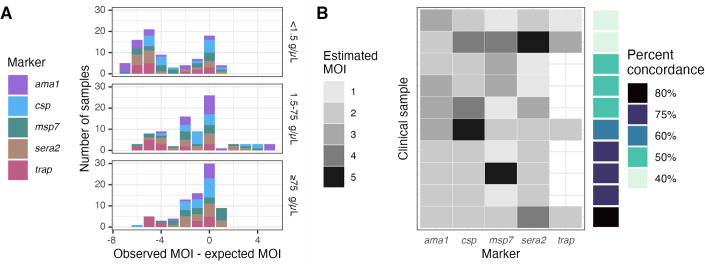
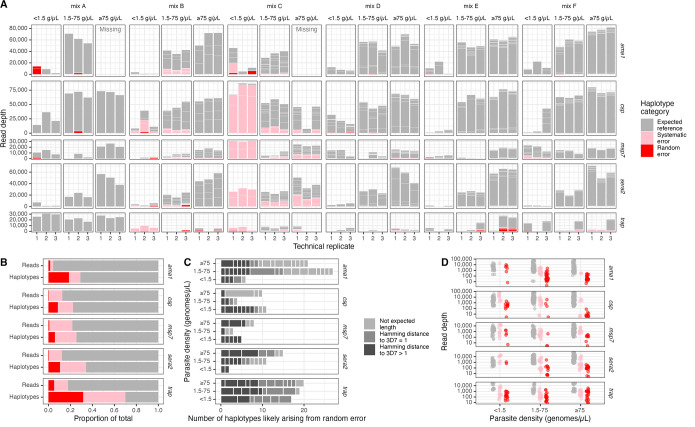
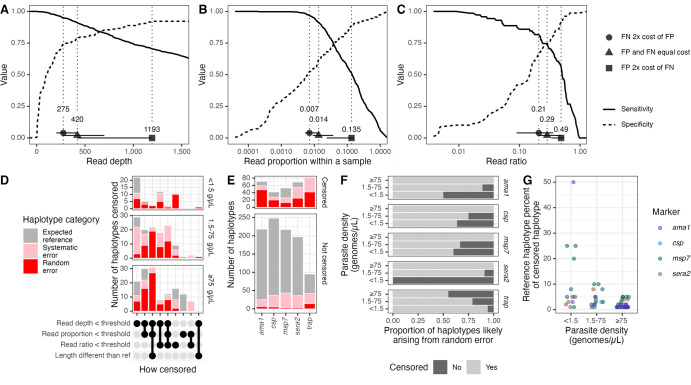
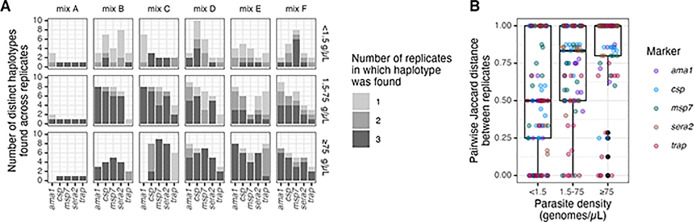
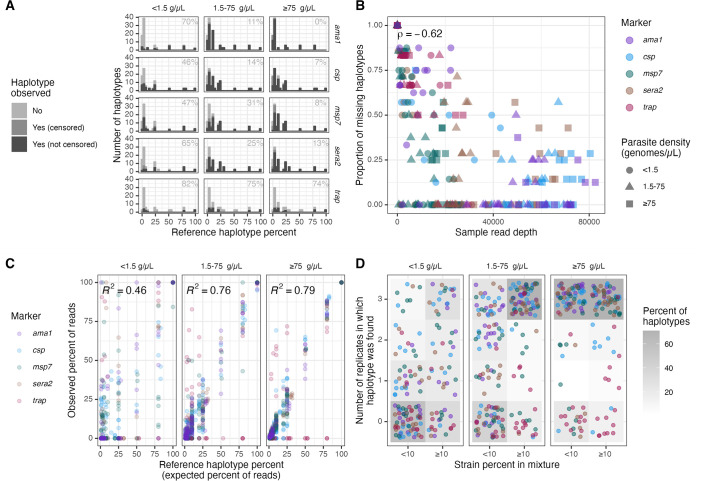
Molecular epidemiologic studies of malaria parasites and other pathogens commonly employ amplicon deep sequencing (AmpSeq) of marker genes derived from dried blood spots (DBS) to answer public health questions related to topics such as transmission and drug resistance. As these methods are increasingly employed to inform direct public health action, it is important to rigorously evaluate the risk of false positive and false negative haplotypes derived from clinically-relevant sample types. We performed a control experiment evaluating haplotype recovery from AmpSeq of 5 marker genes (ama1, csp, msp7, sera2, and trap) from DBS containing mixtures of DNA from 1 to 10 known P. falciparum reference strains across 3 parasite densities in triplicate (n = 270 samples). While false positive haplotypes were present across all parasite densities and mixtures, we optimized censoring criteria to remove 83% (148/179) of false positives while removing only 8% (67/859) of true positives. Post-censoring, the median pairwise Jaccard distance between replicates was 0.83. We failed to recover 35% (477/1365) of haplotypes expected to be present in the sample. Haplotypes were more likely to be missed in low-density samples with <1.5 genomes/μL (OR: 3.88, CI: 1.82-8.27, vs. high-density samples with ≥75 genomes/μL) and in samples with lower read depth (OR per 10,000 reads: 0.61, CI: 0.54-0.69). Furthermore, minority haplotypes within a sample were more likely to be missed than dominant haplotypes (OR per 0.01 increase in proportion: 0.96, CI: 0.96-0.97). Finally, in clinical samples the percent concordance across markers for multiplicity of infection ranged from 40%-80%. Taken together, our observations indicate that, with sufficient read depth, the majority of haplotypes can be successfully recovered from DBS while limiting the false positive rate.
疟原虫和其他病原体的分子流行病学研究通常采用对源自干血斑(DBS)的标记基因进行扩增子深度测序(AmpSeq),以回答与传播和耐药性等主题相关的公共卫生问题。随着这些方法越来越多地用于指导直接的公共卫生行动,严格评估源自临床相关样本类型的假阳性和假阴性单倍型的风险非常重要。我们进行了一项对照实验,评估从含有1至10种已知恶性疟原虫参考菌株DNA混合物的DBS中对5个标记基因(ama1、csp、msp7、sera2和trap)进行AmpSeq时单倍型的恢复情况,实验设置了3种寄生虫密度,每种密度重复3次(n = 270个样本)。虽然在所有寄生虫密度和混合物中都存在假阳性单倍型,但我们优化了审查标准,以去除83%(148/179)的假阳性,同时仅去除8%(67/859)的真阳性。审查后,重复样本之间的中位成对杰卡德距离为0.83。我们未能找回预期存在于样本中的35%(477/1365)的单倍型。在基因组/微升<1.5的低密度样本中,单倍型更有可能被遗漏(与基因组/微升≥75的高密度样本相比,比值比:3.88,置信区间:1.82 - 8.27),并且在读取深度较低的样本中也是如此(每10,000次读取的比值比:0.61,置信区间:0.54 - 0.69)。此外,样本中的少数单倍型比优势单倍型更有可能被遗漏(比例每增加0.01的比值比:0.96,置信区间:0.96 - 0.97)。最后,在临床样本中,不同标记之间感染复数的一致性百分比范围为40% - 80%。综上所述,我们的观察结果表明,在有足够读取深度的情况下,大多数单倍型可以从干血斑中成功找回,同时限制假阳性率。