Center for Biophysics and Quantitative Biology, University of Illinois Urbana-Champaign, Urbana, Illinois, United States of America.
Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory University, Georgia Institute of Technology, Atlanta, Georgia, United States of America.
PLoS Comput Biol. 2024 Jun 5;20(6):e1012103. doi: 10.1371/journal.pcbi.1012103. eCollection 2024 Jun.
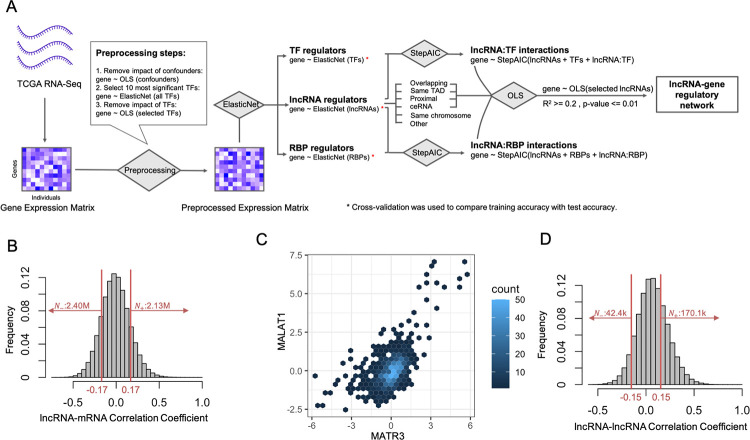
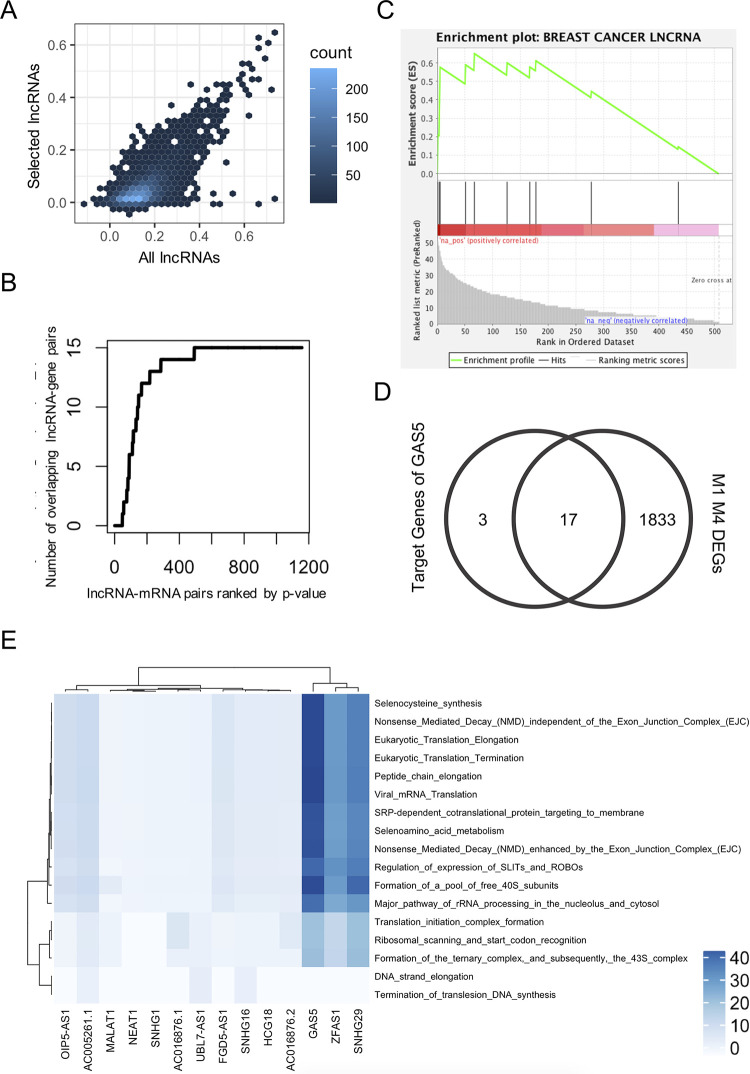
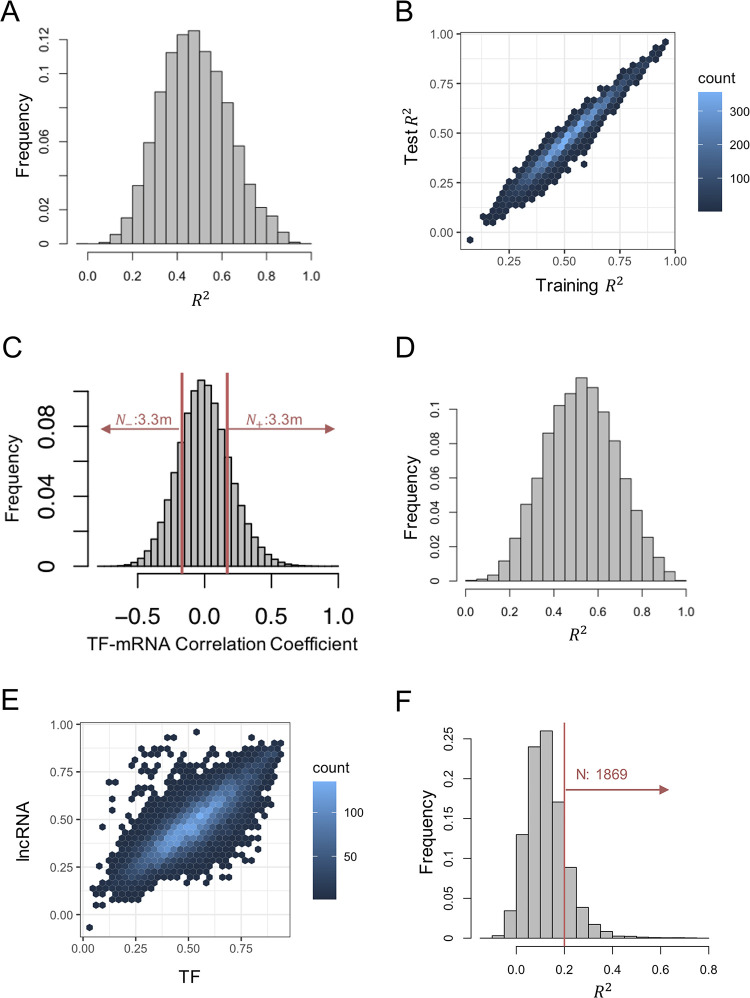
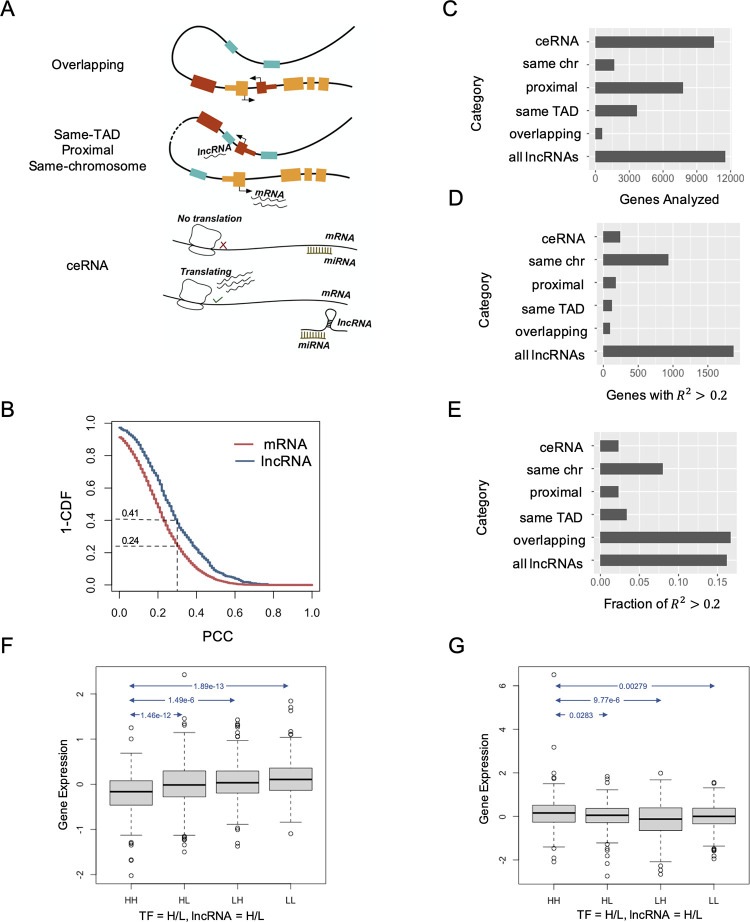
Long non-coding RNAs (lncRNAs) have received attention in recent years for their regulatory roles in diverse biological contexts including cancer, yet large gaps remain in our understanding of their mechanisms and global maps of their targets. In this work, we investigated a basic unanswered question of lncRNA systems biology: to what extent can gene expression variation across individuals be attributed to lncRNA-driven regulation? To answer this, we analyzed RNA-seq data from a cohort of breast cancer patients, explaining each gene's expression variation using a small set of automatically selected lncRNA regulators. A key aspect of this analysis is that it accounts for confounding effects of transcription factors (TFs) as common regulators of a lncRNA-mRNA pair, to enrich the explained gene expression for lncRNA-mediated regulation. We found that for 16% of analyzed genes, lncRNAs can explain more than 20% of expression variation. We observed 25-50% of the putative regulator lncRNAs to be in 'cis' to, i.e., overlapping or located proximally to the target gene. This led us to quantify the global regulatory impact of such cis-located lncRNAs, which was found to be substantially greater than that of trans-located lncRNAs. Additionally, by including statistical interaction terms involving lncRNA-protein pairs as predictors in our regression models, we identified cases where a lncRNA's regulatory effect depends on the presence of a TF or RNA-binding protein. Finally, we created a high-confidence lncRNA-gene regulatory network whose edges are supported by co-expression as well as a plausible mechanism such as cis-action, protein scaffolding or competing endogenous RNAs. Our work is a first attempt to quantify the extent of gene expression control exerted globally by lncRNAs, especially those located proximally to their regulatory targets, in a specific biological (breast cancer) context. It also marks a first step towards systematic reconstruction of lncRNA regulatory networks, going beyond the current paradigm of co-expression networks, and motivates future analyses assessing the generalizability of our findings to additional biological contexts.
长非编码 RNA(lncRNA)在多种生物学背景下(包括癌症)的调控作用近年来受到关注,但我们对其机制和全局靶标的了解仍存在很大差距。在这项工作中,我们研究了 lncRNA 系统生物学的一个基本未解决的问题:个体间的基因表达变化在多大程度上可以归因于 lncRNA 驱动的调节?为了回答这个问题,我们分析了一组乳腺癌患者的 RNA-seq 数据,使用一小部分自动选择的 lncRNA 调节剂来解释每个基因的表达变化。该分析的一个关键方面是,它考虑了转录因子(TF)作为 lncRNA-mRNA 对共同调节剂的混杂效应,以丰富 lncRNA 介导的调节的解释基因表达。我们发现,对于分析的 16%的基因,lncRNA 可以解释超过 20%的表达变化。我们观察到 25-50%的假定调节剂 lncRNA 位于“顺式”位置,即与靶基因重叠或位于靶基因附近。这使我们能够量化这种顺式定位 lncRNA 的全局调节作用,发现其作用明显大于反式定位 lncRNA 的作用。此外,通过在回归模型中包含涉及 lncRNA-蛋白质对的统计相互作用项作为预测因子,我们确定了 lncRNA 的调节作用取决于 TF 或 RNA 结合蛋白存在的情况。最后,我们创建了一个高可信度的 lncRNA-基因调控网络,其边缘由共表达以及合理的机制(如顺式作用、蛋白质支架或竞争内源性 RNA)支持。我们的工作首次尝试定量地确定 lncRNA 在特定生物学(乳腺癌)背景下全局控制基因表达的程度,特别是那些位于其调节靶标附近的 lncRNA。它还标志着系统地重建 lncRNA 调控网络的第一步,超越了当前的共表达网络范例,并激励未来的分析评估我们的发现对其他生物学背景的普遍性。