Department of Advanced Computing Sciences, Maastricht University, Maastricht, The Netherlands.
MAP5, Université Paris Cité, CNRS, Paris, France.
Bioinformatics. 2024 Jun 3;40(6). doi: 10.1093/bioinformatics/btae337.
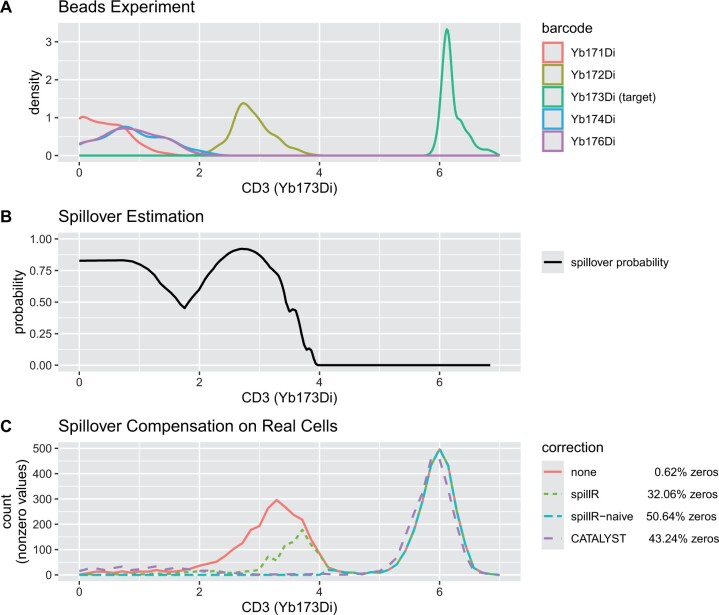
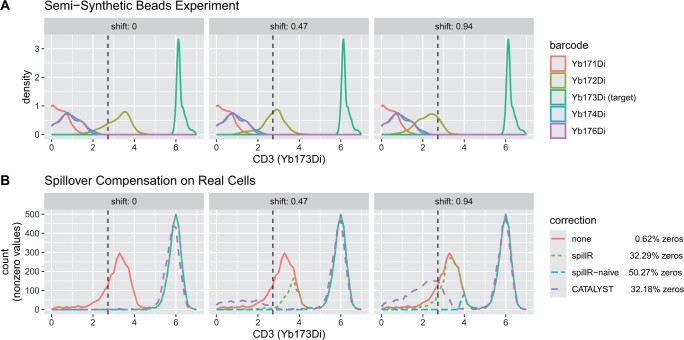
Channel interference in mass cytometry can cause spillover and may result in miscounting of protein markers. Chevrier et al. introduce an experimental and computational procedure to estimate and compensate for spillover implemented in their R package CATALYST. They assume spillover can be described by a spillover matrix that encodes the ratio between the signal in the unstained spillover receiving and stained spillover emitting channel. They estimate the spillover matrix from experiments with beads. We propose to skip the matrix estimation step and work directly with the full bead distributions. We develop a nonparametric finite mixture model and use the mixture components to estimate the probability of spillover. Spillover correction is often a pre-processing step followed by downstream analyses, and choosing a flexible model reduces the chance of introducing biases that can propagate downstream.
We implement our method in an R package spillR using expectation-maximization to fit the mixture model. We test our method on simulated, semi-simulated, and real data from CATALYST. We find that our method compensates low counts accurately, does not introduce negative counts, avoids overcompensating high counts, and preserves correlations between markers that may be biologically meaningful.
Our new R package spillR is on bioconductor at bioconductor.org/packages/spillR. All experiments and plots can be reproduced by compiling the R markdown file spillR_paper.Rmd at github.com/ChristofSeiler/spillR_paper.
在液滴数字流式细胞术中,通道干扰可能导致串扰,并可能导致蛋白标记物的计数错误。Chevrier 等人引入了一种实验和计算程序,以估计和补偿他们的 R 包 CATALYST 中实现的串扰。他们假设串扰可以用一个溢出矩阵来描述,该矩阵编码了未染色的溢出接收通道和染色的溢出发射通道之间的信号比值。他们从珠子实验中估计溢出矩阵。我们建议跳过矩阵估计步骤,直接使用完整的珠子分布。我们开发了一个非参数有限混合模型,并使用混合成分来估计串扰的概率。串扰校正通常是下游分析之前的预处理步骤,选择灵活的模型可以减少引入下游传播的偏差的机会。
我们使用期望最大化在 R 包 spillR 中实现了我们的方法,以拟合混合模型。我们在模拟、半模拟和来自 CATALYST 的真实数据上测试了我们的方法。我们发现我们的方法可以准确地补偿低计数,不会引入负计数,避免过度补偿高计数,并保留可能具有生物学意义的标记之间的相关性。
我们的新 R 包 spillR 可在 bioconductor.org/packages/spillR 上获得。通过编译 R markdown 文件 spillR_paper.Rmd 在 github.com/ChristofSeiler/spillR_paper 上可以重现所有的实验和图。