Quantitative Virology and Evolution Unit, Laboratory of Viral Diseases, NIAID, NIH, Bethesda, MD, USA.
Sci Adv. 2024 Jun 21;10(25):eado1693. doi: 10.1126/sciadv.ado1693. Epub 2024 Jun 19.
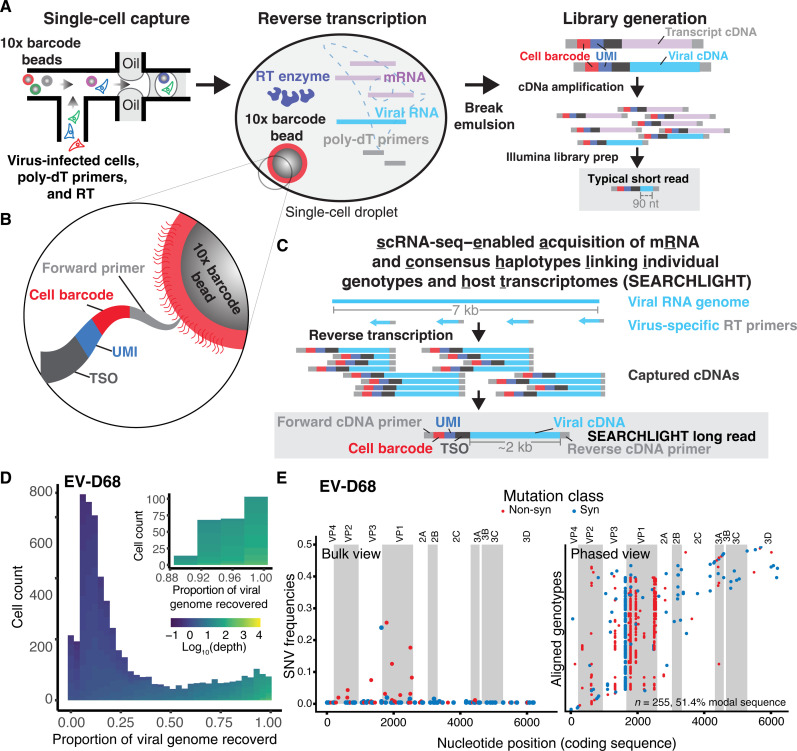
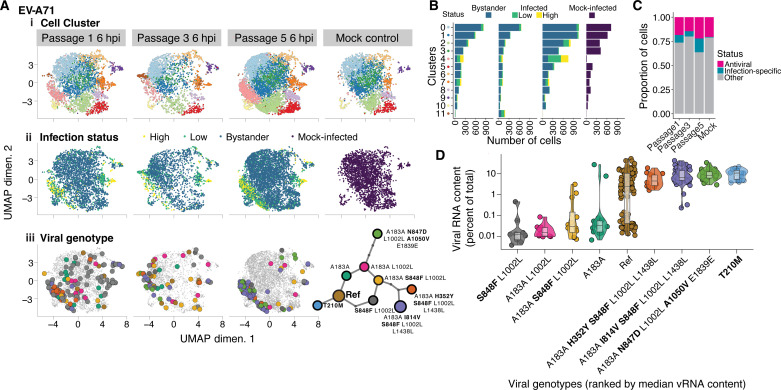
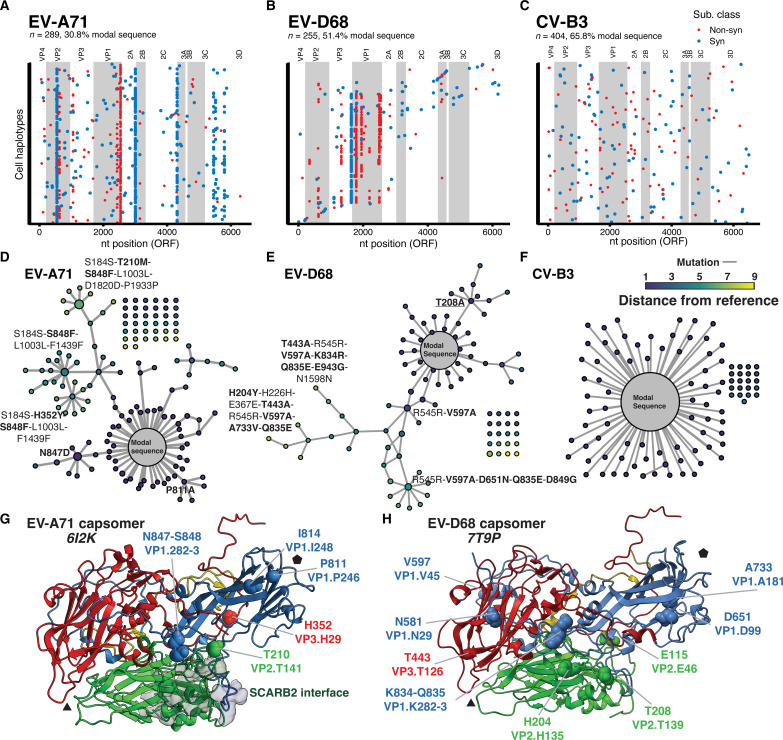
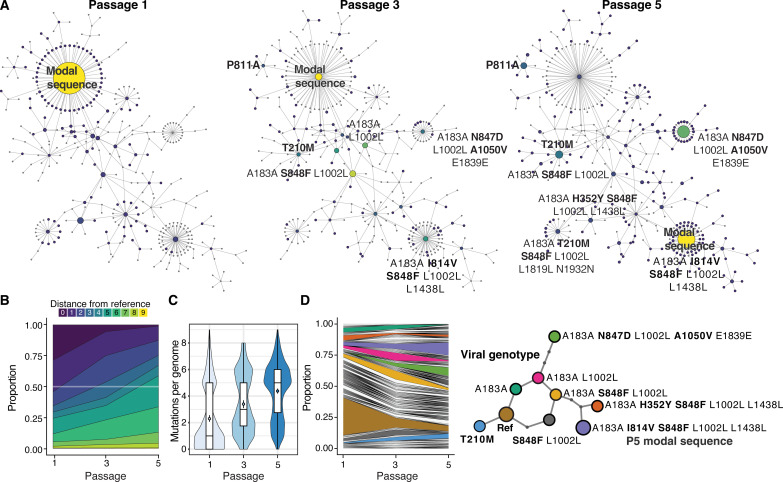
Like all biological populations, viral populations exist as networks of genotypes connected through mutation. Mapping the topology of these networks and quantifying population dynamics across them is crucial to understanding how populations adapt to changes in their selective environment. The influence of mutational networks is especially profound in viral populations that rapidly explore their mutational neighborhoods via high mutation rates. Using a single-cell sequencing method, scRNA-seq-enabled acquisition of mRNA and consensus haplotypes linking individual genotypes and host transcriptomes (SEARCHLIGHT), we captured and assembled viral haplotypes from hundreds of individual infected cells, revealing the complexity of viral population structures. We obtained these genotypes in parallel with host cell transcriptome information, enabling us to link host cell transcriptional phenotypes to the genetic structures underlying virus adaptation. Our examination of these structures reveals the common evolutionary dynamics of enterovirus populations and illustrates how viral populations reach through mutational "tunnels" to span evolutionary landscapes and maintain connection with multiple adaptive genotypes simultaneously.
与所有生物种群一样,病毒种群作为通过突变连接的基因型网络而存在。绘制这些网络的拓扑结构并量化它们之间的种群动态对于理解种群如何适应选择环境的变化至关重要。在通过高突变率快速探索突变邻域的病毒种群中,突变网络的影响尤为深远。使用单细胞测序方法,即 scRNA-seq 实现的与个体基因型和宿主转录组相关联的 mRNA 和共识单倍型的获取(SEARCHLIGHT),我们从数百个受感染的细胞中捕获并组装了病毒单倍型,揭示了病毒种群结构的复杂性。我们在获得这些基因型的同时获取了宿主细胞转录组信息,使我们能够将宿主细胞转录表型与病毒适应的遗传结构联系起来。我们对这些结构的研究揭示了肠道病毒种群的常见进化动态,并说明了病毒种群如何通过突变“隧道”跨越进化景观并同时与多个适应性基因型保持连接。