Department of Microbiology and Immunology, Stanford University School of Medicine, Stanford, CA, USA.
Department of Chemical and Systems Biology, Chem-H and Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA, USA.
Nat Commun. 2024 Jun 19;15(1):5179. doi: 10.1038/s41467-024-49161-9.
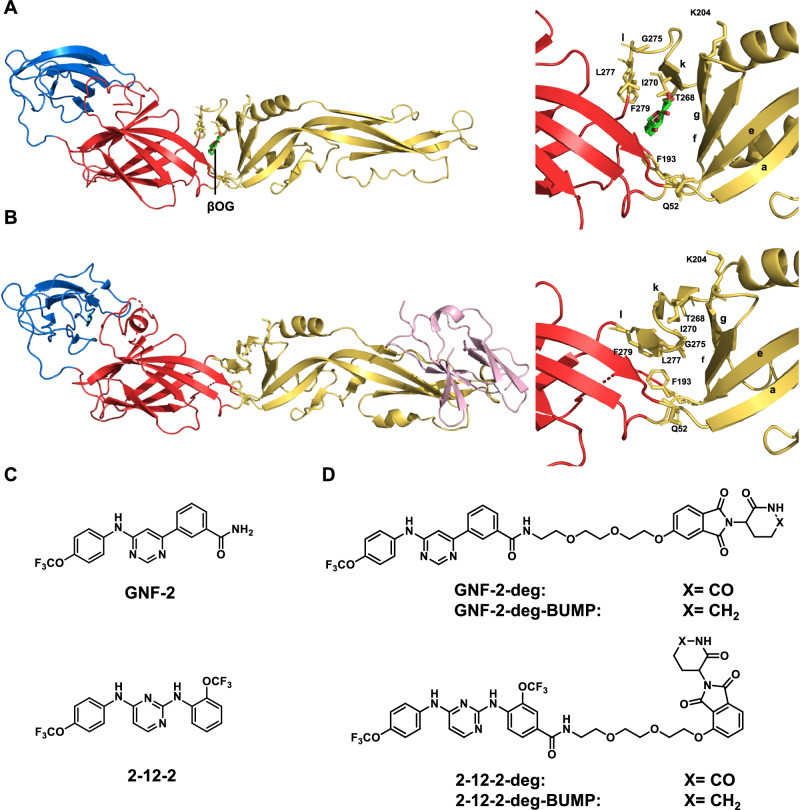
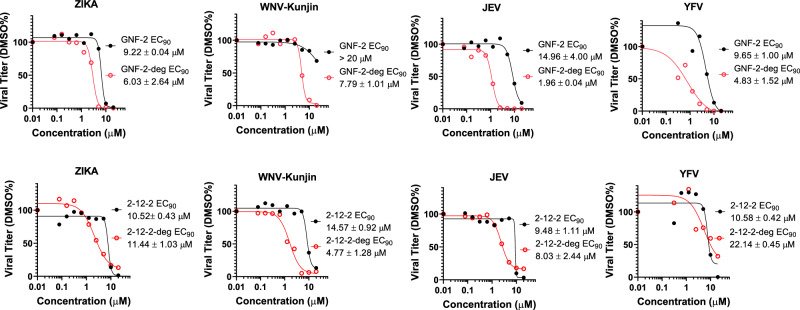
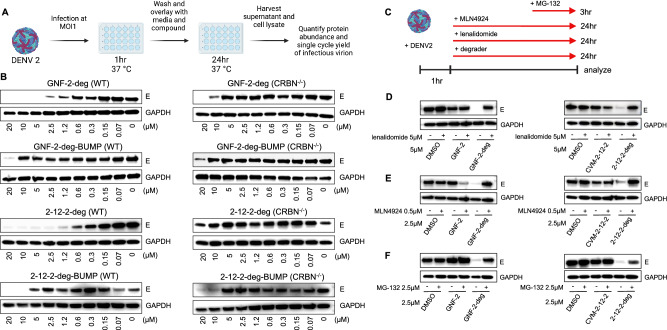
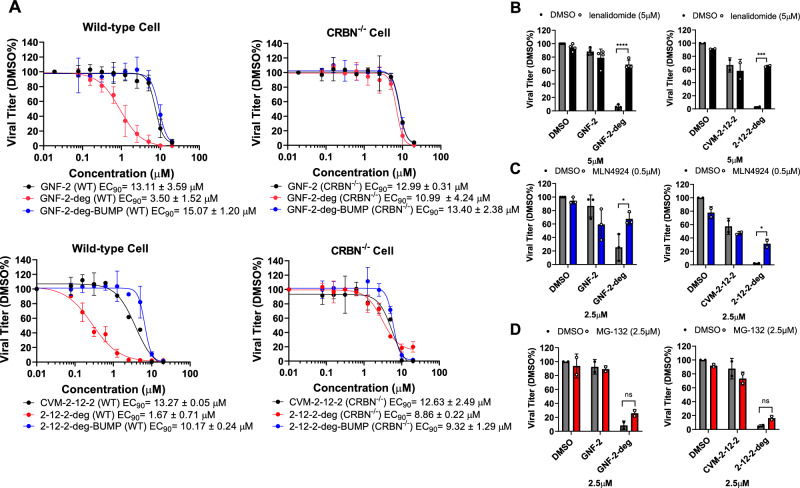
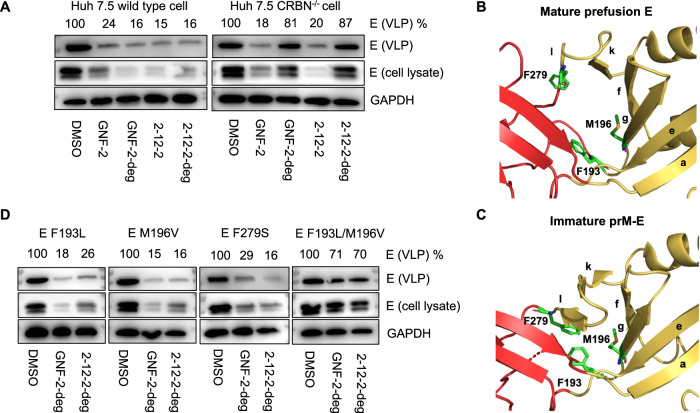
Viral genetic diversity presents significant challenges in developing antivirals with broad-spectrum activity and high barriers to resistance. Here we report development of proteolysis targeting chimeras (PROTACs) targeting the dengue virus envelope (E) protein through coupling of known E fusion inhibitors to ligands of the CRL4 E3 ubiquitin ligase. The resulting small molecules block viral entry through inhibition of E-mediated membrane fusion and interfere with viral particle production by depleting intracellular E in infected Huh 7.5 cells. This activity is retained in the presence of point mutations previously shown to confer partial resistance to the parental inhibitors due to decreased inhibitor-binding. The E PROTACs also exhibit broadened spectrum of activity compared to the parental E inhibitors against a panel of mosquito-borne flaviviruses. These findings encourage further exploration of targeted protein degradation as a differentiated and potentially advantageous modality for development of broad-spectrum direct-acting antivirals.
病毒遗传多样性给广谱活性和高耐药性的抗病毒药物的开发带来了重大挑战。在这里,我们报告了通过将已知的 E 融合抑制剂与 CRL4 E3 泛素连接酶的配体偶联,针对登革热病毒包膜 (E) 蛋白开发蛋白酶靶向嵌合体 (PROTACs)。这些小分子通过抑制 E 介导的膜融合来阻止病毒进入,并通过耗尽感染的 Huh 7.5 细胞内的 E 来干扰病毒颗粒的产生。由于抑制剂结合减少,该活性在先前显示对亲本抑制剂产生部分耐药性的点突变存在的情况下得以保留。与亲本 E 抑制剂相比,E PROTACs 对一组蚊媒黄病毒也表现出更广泛的活性谱。这些发现鼓励进一步探索靶向蛋白降解作为开发广谱直接作用抗病毒药物的一种差异化和潜在有利的模式。