Institute of Genomics and RNomics, Biocenter Innsbruck, Medical University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria.
Institute of Genomics and RNomics, Biocenter Innsbruck, Medical University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria.
Am J Hum Genet. 2024 Jul 11;111(7):1383-1404. doi: 10.1016/j.ajhg.2024.05.023. Epub 2024 Jun 21.
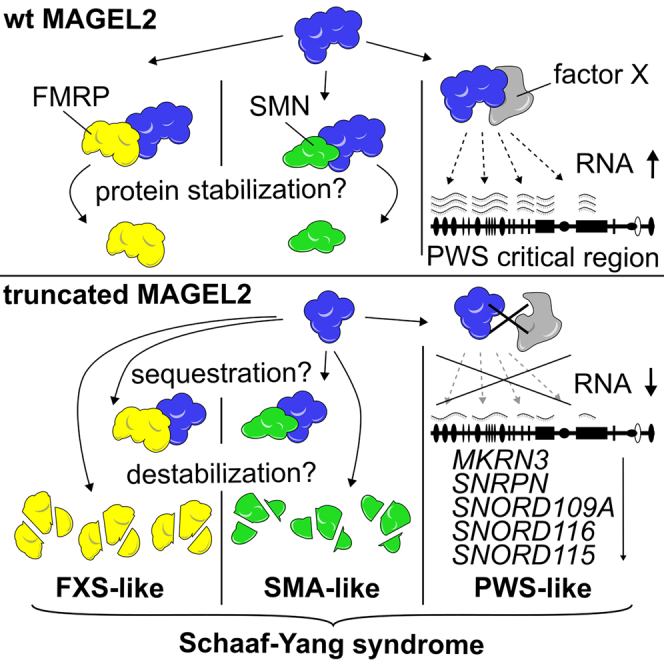
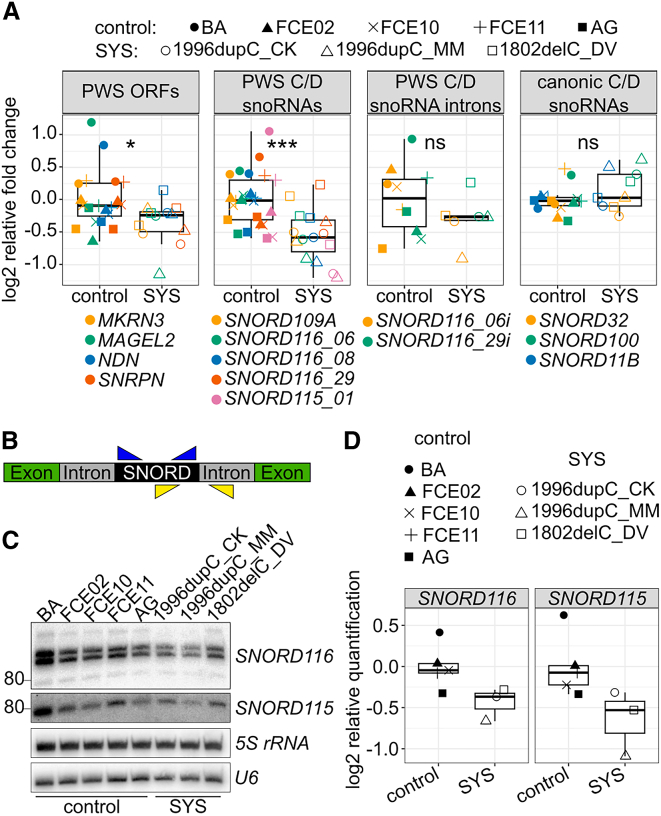
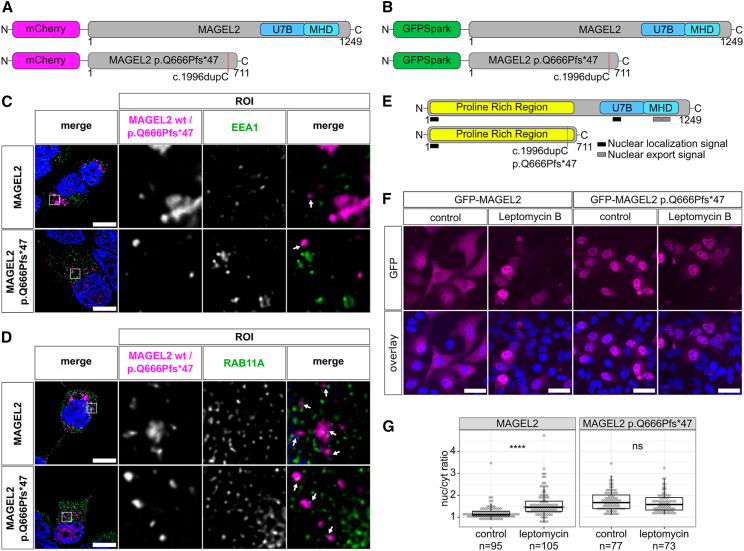
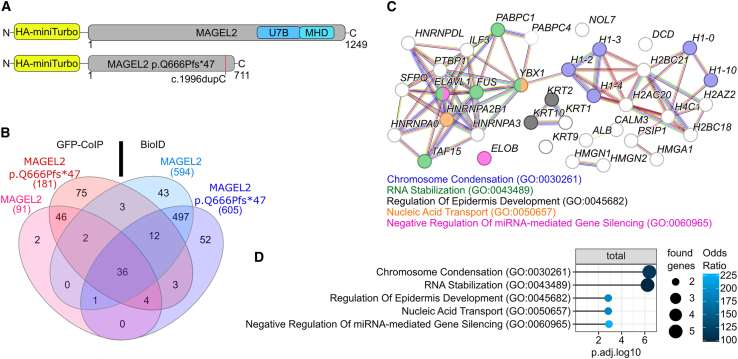
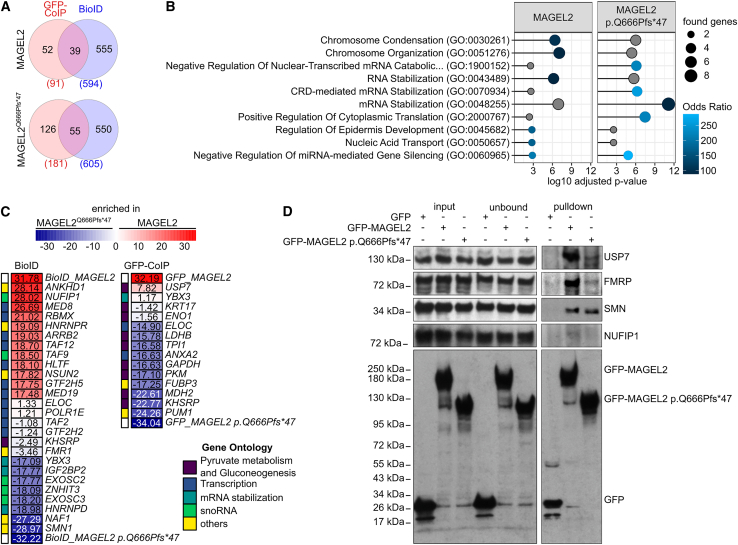
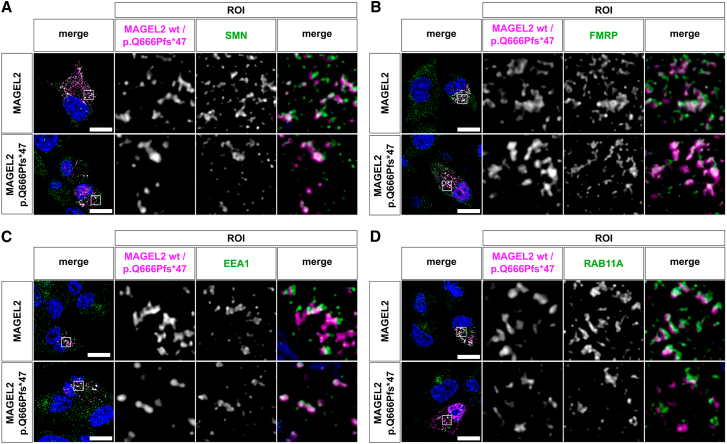
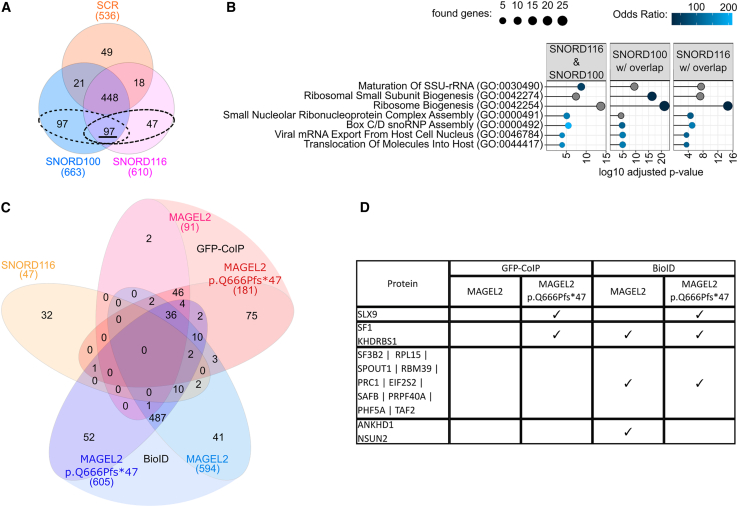
The neurodevelopmental disorders Prader-Willi syndrome (PWS) and Schaaf-Yang syndrome (SYS) both arise from genomic alterations within human chromosome 15q11-q13. A deletion of the SNORD116 cluster, encoding small nucleolar RNAs, or frameshift mutations within MAGEL2 result in closely related phenotypes in individuals with PWS or SYS, respectively. By investigation of their subcellular localization, we observed that in contrast to a predominant cytoplasmic localization of wild-type (WT) MAGEL2, a truncated MAGEL2 mutant was evenly distributed between the cytoplasm and the nucleus. To elucidate regulatory pathways that may underlie both diseases, we identified protein interaction partners for WT or mutant MAGEL2, in particular the survival motor neuron protein (SMN), involved in spinal muscular atrophy, and the fragile-X-messenger ribonucleoprotein (FMRP), involved in autism spectrum disorders. The interactome of the non-coding RNA SNORD116 was also investigated by RNA-CoIP. We show that WT and truncated MAGEL2 were both involved in RNA metabolism, while regulation of transcription was mainly observed for WT MAGEL2. Hence, we investigated the influence of MAGEL2 mutations on the expression of genes from the PWS locus, including the SNORD116 cluster. Thereby, we provide evidence for MAGEL2 mutants decreasing the expression of SNORD116, SNORD115, and SNORD109A, as well as protein-coding genes MKRN3 and SNRPN, thus bridging the gap between PWS and SYS.
神经发育障碍普拉德-威利综合征(PWS)和 Schaaf-Yang 综合征(SYS)均源自人类 15q11-q13 染色体上的基因组改变。SNORD116 簇的缺失,或 MAGEL2 内的移码突变,分别导致 PWS 或 SYS 个体具有密切相关的表型。通过对其亚细胞定位的研究,我们观察到与野生型(WT)MAGEL2 主要定位于细胞质相反,截断的 MAGEL2 突变体在细胞质和细胞核之间均匀分布。为了阐明可能导致这两种疾病的调节途径,我们鉴定了 WT 或突变型 MAGEL2 的蛋白相互作用伙伴,特别是参与脊髓性肌萎缩症的存活运动神经元蛋白(SMN)和参与自闭症谱系障碍的脆性 X 信使核糖核蛋白(FMRP)。非编码 RNA SNORD116 的相互作用组也通过 RNA-CoIP 进行了研究。我们表明,WT 和截断的 MAGEL2 均参与 RNA 代谢,而转录的调节主要观察到 WT MAGEL2。因此,我们研究了 MAGEL2 突变对 PWS 基因座包括 SNORD116 簇的基因表达的影响。由此,我们提供了 MAGEL2 突变体降低 SNORD116、SNORD115 和 SNORD109A 以及编码蛋白的基因 MKRN3 和 SNRPN 表达的证据,从而弥合了 PWS 和 SYS 之间的差距。