Center for Artificial Intelligence Research, Wake Forest University School of Medicine, Winston-Salem, NC 27101, USA.
Division of Medical Oncology, James Cancer Hospital and the Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, USA.
Int J Mol Sci. 2024 Jul 3;25(13):7306. doi: 10.3390/ijms25137306.
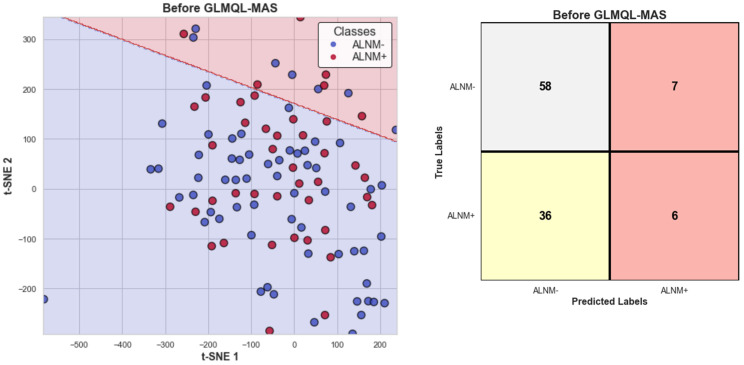
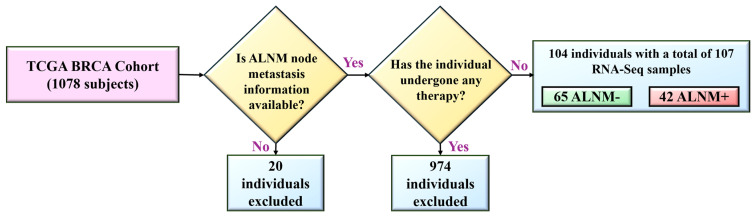
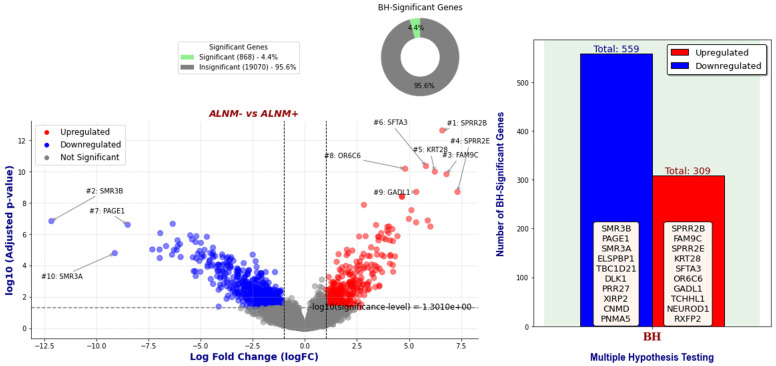
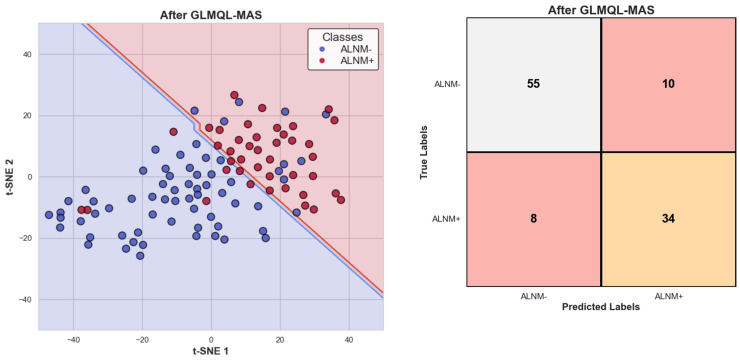
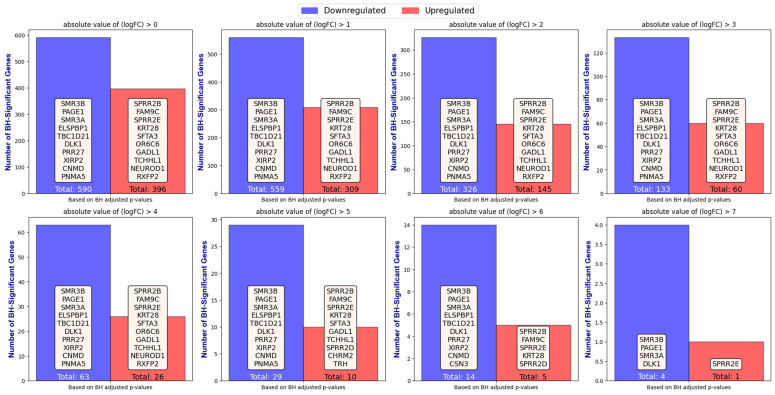
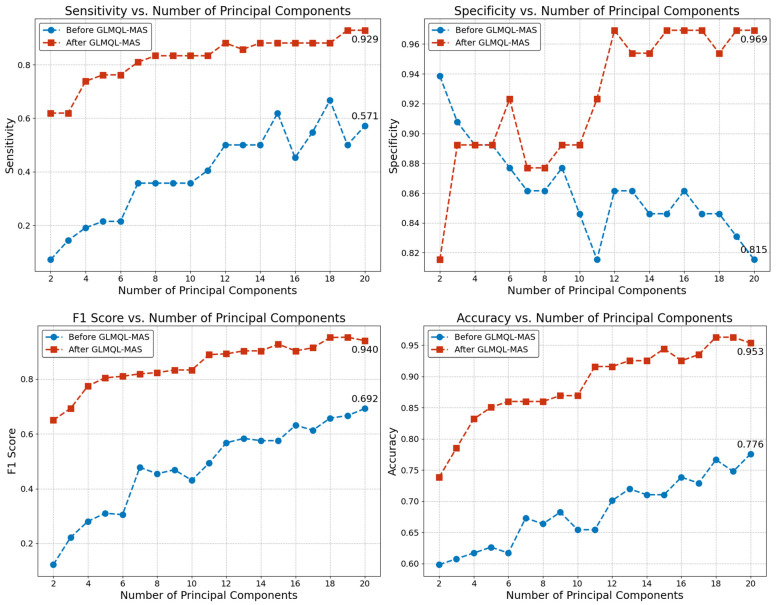
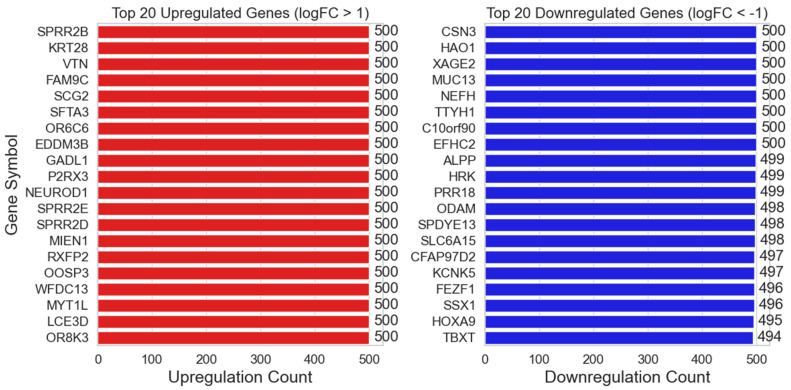
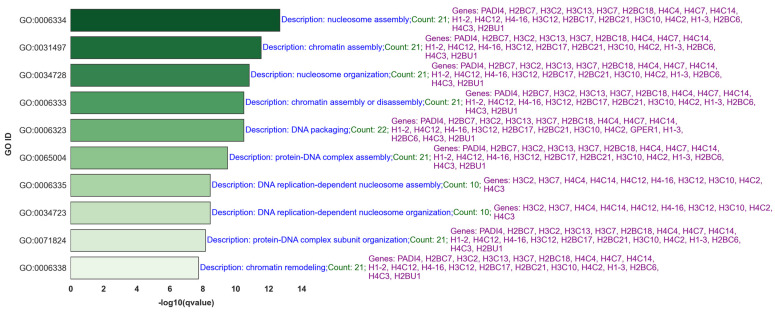
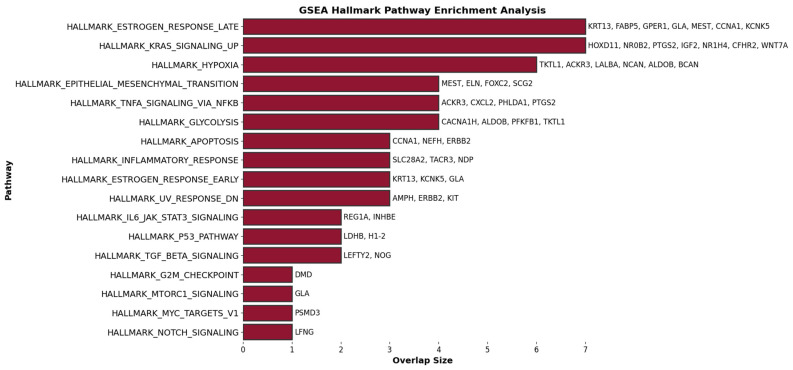
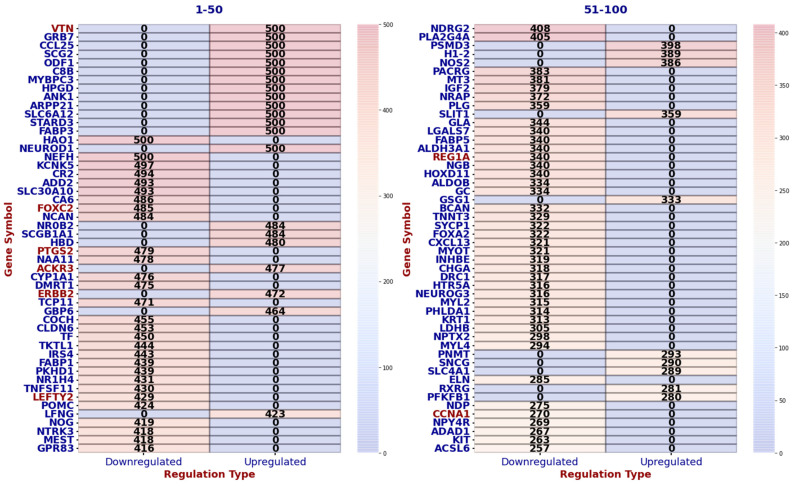
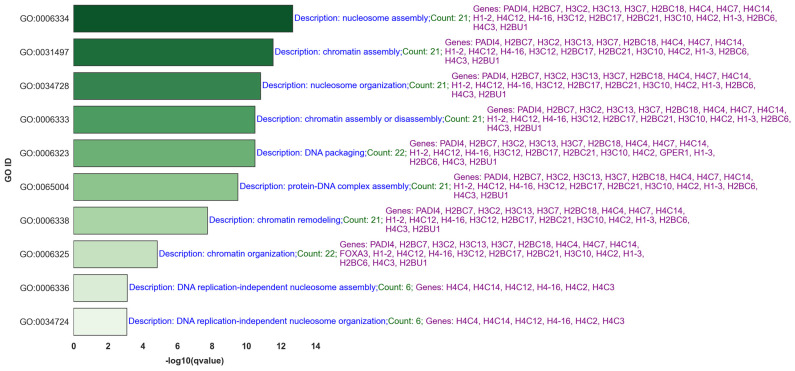
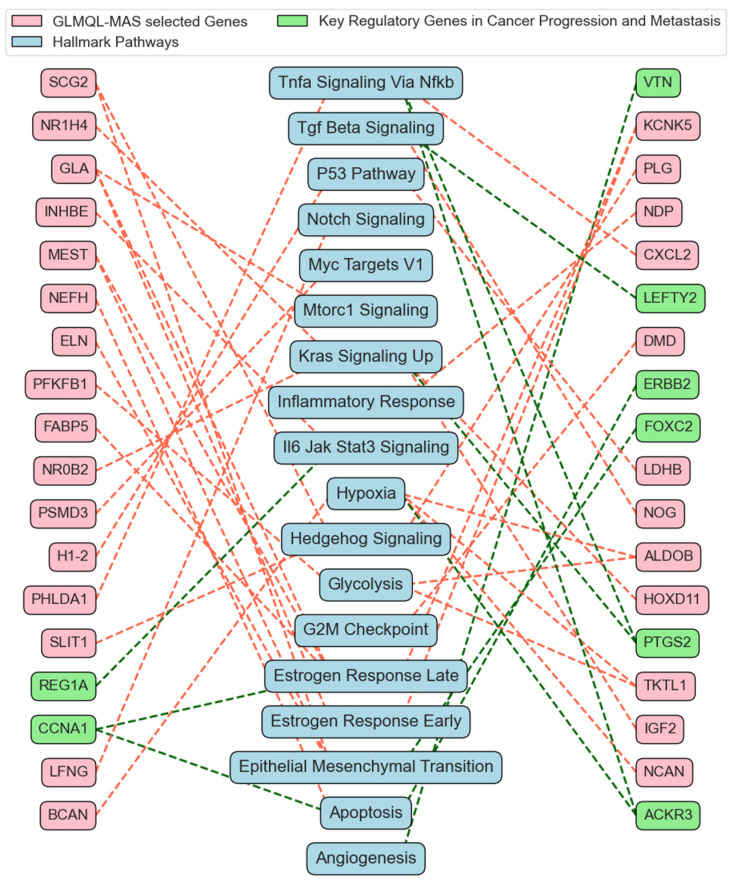
Our study aims to address the methodological challenges frequently encountered in RNA-Seq data analysis within cancer studies. Specifically, it enhances the identification of key genes involved in axillary lymph node metastasis (ALNM) in breast cancer. We employ Generalized Linear Models with Quasi-Likelihood (GLMQLs) to manage the inherently discrete and overdispersed nature of RNA-Seq data, marking a significant improvement over conventional methods such as the -test, which assumes a normal distribution and equal variances across samples. We utilize the Trimmed Mean of M-values (TMMs) method for normalization to address library-specific compositional differences effectively. Our study focuses on a distinct cohort of 104 untreated patients from the TCGA Breast Invasive Carcinoma (BRCA) dataset to maintain an untainted genetic profile, thereby providing more accurate insights into the genetic underpinnings of lymph node metastasis. This strategic selection paves the way for developing early intervention strategies and targeted therapies. Our analysis is exclusively dedicated to protein-coding genes, enriched by the Magnitude Altitude Scoring (MAS) system, which rigorously identifies key genes that could serve as predictors in developing an ALNM predictive model. Our novel approach has pinpointed several genes significantly linked to ALNM in breast cancer, offering vital insights into the molecular dynamics of cancer development and metastasis. These genes, including , , , , , , and , are involved in key processes like apoptosis, epithelial-mesenchymal transition, angiogenesis, response to hypoxia, and KRAS signaling pathways, which are crucial for tumor virulence and the spread of metastases. Moreover, the approach has also emphasized the importance of the small proline-rich protein family (SPRR), including , , and , recognized for their significant involvement in cancer-related pathways and their potential as therapeutic targets. Important transcripts such as , , , and others have been highlighted as critical in modulating the chromatin structure and gene expression, fundamental for the progression and spread of cancer.
我们的研究旨在解决癌症研究中 RNA-Seq 数据分析中经常遇到的方法学挑战。具体来说,它增强了对乳腺癌腋窝淋巴结转移 (ALNM) 中涉及的关键基因的识别。我们使用广义线性模型与拟似然 (GLMQL) 来处理 RNA-Seq 数据固有的离散性和过度分散性,这比传统方法如 -检验有了显著的改进,-检验假设数据分布正态且样本间方差相等。我们使用均一化值的 trimmed mean (TMM) 方法来有效解决文库特异性组成差异问题。我们的研究集中在 TCGA 乳腺癌浸润性癌 (BRCA) 数据集的 104 名未经治疗的患者的独特队列上,以保持未受污染的遗传特征,从而更准确地了解淋巴结转移的遗传基础。这种策略性选择为开发早期干预策略和靶向治疗铺平了道路。我们的分析专门针对蛋白质编码基因进行,这些基因通过 Magnitude Altitude Scoring (MAS) 系统进行富集,该系统严格识别出可能作为开发 ALNM 预测模型的预测因子的关键基因。我们的新方法确定了几个与乳腺癌中 ALNM 显著相关的基因,为癌症发展和转移的分子动力学提供了重要的见解。这些基因包括、、、、、和,它们参与了关键过程,如细胞凋亡、上皮-间充质转化、血管生成、缺氧反应和 KRAS 信号通路,这些过程对于肿瘤毒力和转移的扩散至关重要。此外,该方法还强调了小富含脯氨酸蛋白家族 (SPRR) 的重要性,包括、和,它们在癌症相关途径中具有显著的参与度,并且可能成为治疗靶点。重要的转录物如、、和其他转录物被强调为调节染色质结构和基因表达的关键,这对于癌症的进展和扩散至关重要。