Zhuang Beryl C, Jude Marcia Smiti, Konwar Chaini, Yusupov Natan, Ryan Calen P, Engelbrecht Hannah-Ruth, Whitehead Joanne, Halberstam Alexandra A, MacIsaac Julia L, Dever Kristy, Tran Toan Khanh, Korinek Kim, Zimmer Zachary, Lee Nanette R, McDade Thomas W, Kuzawa Christopher W, Huffman Kim M, Belsky Daniel W, Binder Elisabeth B, Czamara Darina, Korthauer Keegan, Kobor Michael S
BC Children's Hospital Research Institute, 950 West 28th Avenue, Vancouver, BC, V5Z 4H4, Canada.
Department of Medical Genetics, Faculty of Medicine, University of British Columbia, Vancouver, BC, V6T 1Z3, Canada.
bioRxiv. 2024 Sep 28:2024.07.02.600461. doi: 10.1101/2024.07.02.600461.
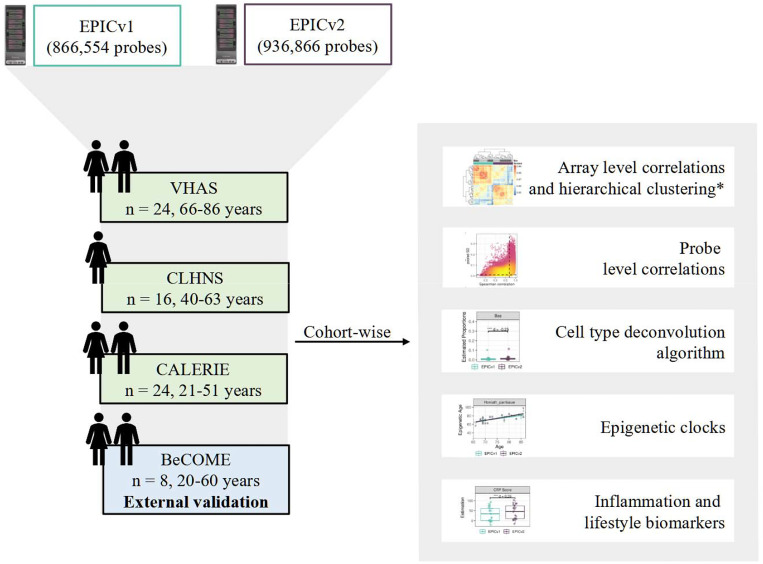
The recently launched DNA methylation profiling platform, Illumina MethylationEPIC BeadChip Infinium microarray v2.0 (EPICv2), is highly correlated with measurements obtained from its predecessor MethylationEPIC BeadChip Infinium microarray v1.0 (EPICv1). However, the concordance between the two versions in the context of DNA methylation-based tools, including cell type deconvolution algorithms, epigenetic clocks, and inflammation and lifestyle biomarkers has not yet been investigated. To address this, we profiled DNA methylation on both EPIC versions using matched venous blood samples from individuals spanning early to late adulthood across four cohorts.
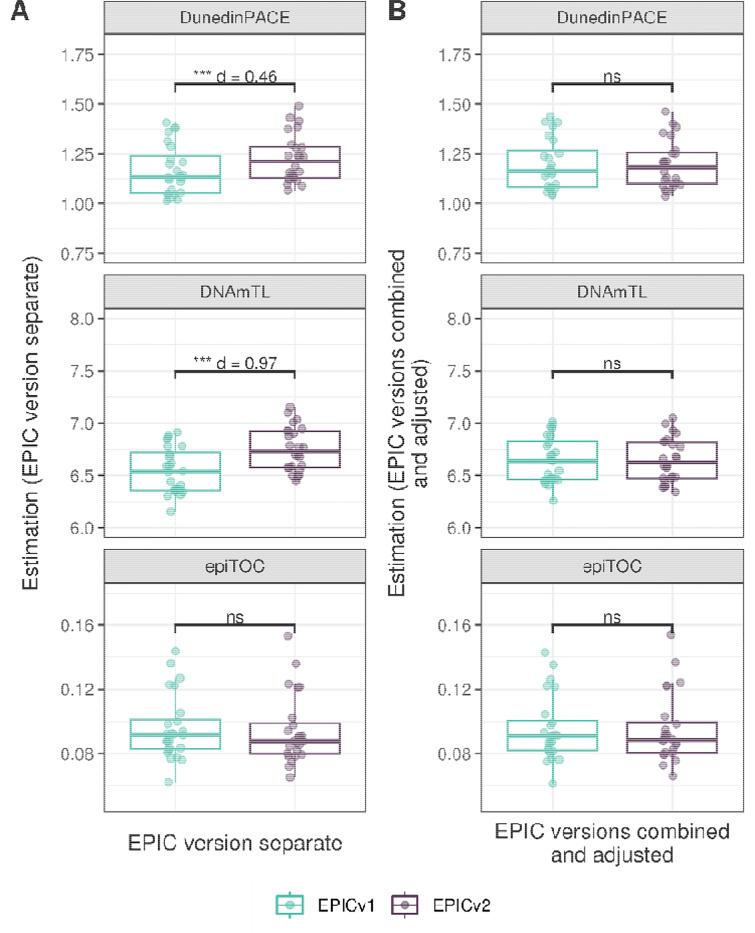
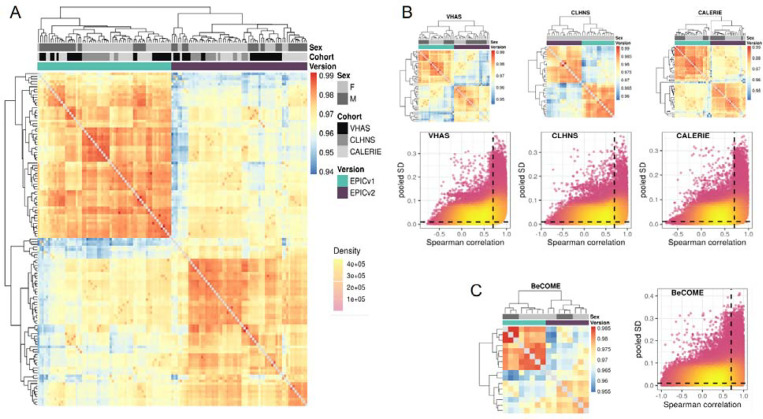
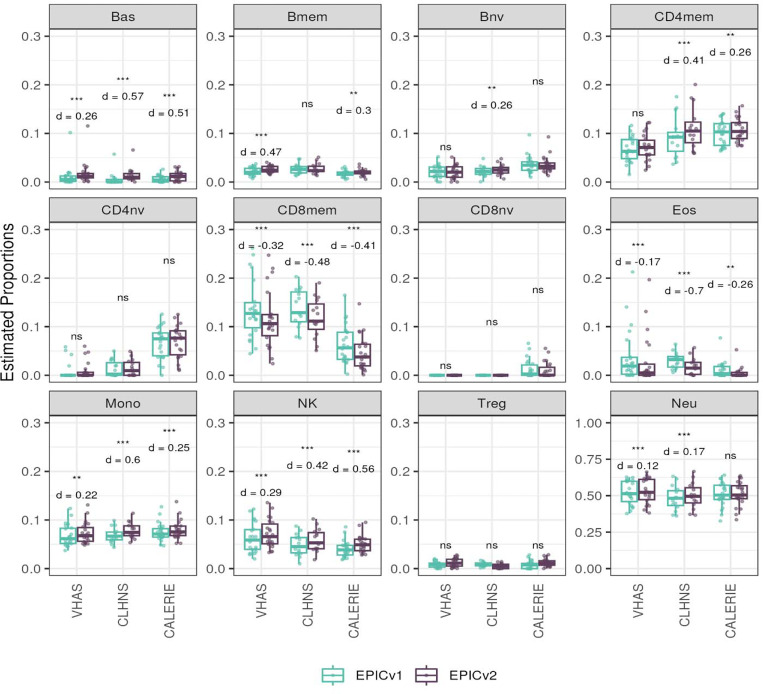
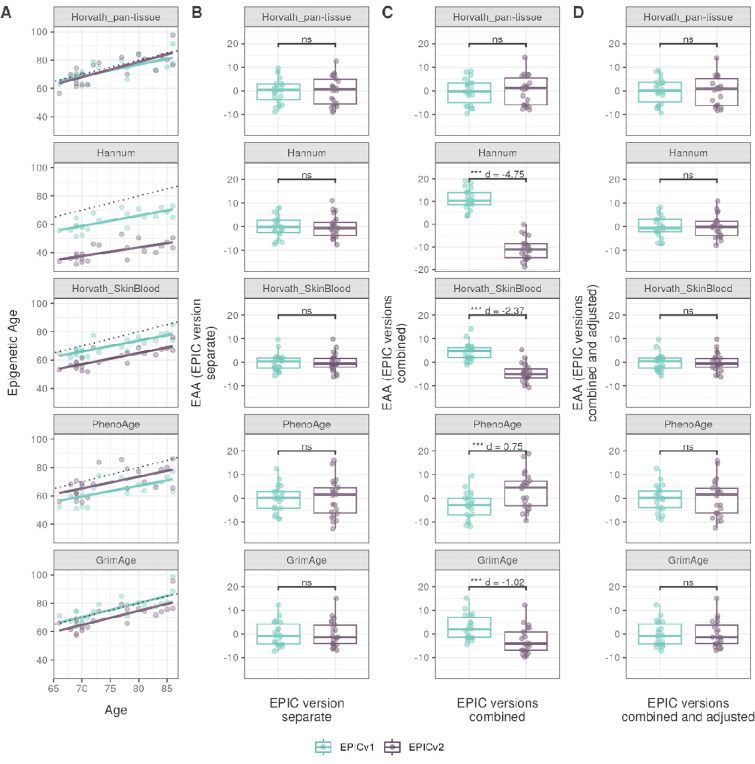
Within each cohort, samples primarily clustered by the EPIC version they were measured on. High concordance between EPIC versions at the array level, but variable concordance at the individual probe level was noted. Significant differences between versions in estimates from DNA methylation-based tools were observed, irrespective of the normalization method, with some nuanced differences across cohorts and tools. Adjusting for EPIC version or calculating estimates separately for each version largely mitigated these version-specific discordances.
Our work illustrates the importance of accounting for EPIC version differences in research scenarios, especially in meta-analyses and longitudinal studies, when samples profiled across different versions are harmonized. Alongside DNA methylation-based tools, our observations also have implications in interpretation of epigenome-wide association studies (EWAS) findings, when results obtained from one version are compared to another, particularly for probes that are poorly concordant between versions.
最近推出的DNA甲基化分析平台Illumina MethylationEPIC BeadChip Infinium微阵列v2.0(EPICv2)与其前身MethylationEPIC BeadChip Infinium微阵列v1.0(EPICv1)所获得的测量结果高度相关。然而,在基于DNA甲基化的工具(包括细胞类型反卷积算法、表观遗传时钟以及炎症和生活方式生物标志物)方面,这两个版本之间的一致性尚未得到研究。为了解决这一问题,我们使用来自四个队列中从成年早期到晚期的个体的匹配静脉血样本,对两个EPIC版本的DNA甲基化进行了分析。
在每个队列中,样本主要根据其测量所使用的EPIC版本进行聚类。在阵列水平上,EPIC版本之间具有高度一致性,但在单个探针水平上一致性存在差异。无论采用何种归一化方法,基于DNA甲基化的工具的估计值在版本之间均观察到显著差异,不同队列和工具之间存在一些细微差别。对EPIC版本进行调整或分别为每个版本计算估计值在很大程度上减轻了这些特定于版本的不一致性。
我们的工作说明了在研究场景中考虑EPIC版本差异的重要性,特别是在对不同版本分析的样本进行整合的荟萃分析和纵向研究中。除了基于DNA甲基化的工具外,我们的观察结果对于表观基因组范围关联研究(EWAS)结果的解释也具有启示意义,即在将一个版本的结果与另一个版本的结果进行比较时,特别是对于版本之间一致性较差的探针。