Elhassan Elhussein A E, Kmochová Tereza, Benson Katherine A, Fennelly Neil K, Barešová Veronika, Kidd Kendrah, Doyle Brendan, Dorman Anthony, Morrin Martina M, Kyne Niamh C, Vyleťal Petr, Hartmannová Hana, Hodaňová Kateřina, Sovová Jana, Mušálková Dita, Vrbacká Alena, Přistoupilová Anna, Živný Jan, Svojšová Klára, Radina Martin, Stránecký Viktor, Loginov Dmitry, Pompach Petr, Novák Petr, Vaníčková Zdislava, Hansíková Hana, Rajnochová-Bloudíčková Silvie, Viklický Ondřej, Hůlková Helena, Cavalleri Gianpiero L, Hnízda Aleš, Bleyer Anthony J, Kmoch Stanislav, Conlon Peter J, Živná Martina
Department of Nephrology and Transplantation, Beaumont Hospital, Dublin, Ireland.
Department of Medicine, Royal College of Surgeons in Ireland, Dublin, Ireland.
Kidney Int Rep. 2024 Apr 15;9(7):2209-2226. doi: 10.1016/j.ekir.2024.04.031. eCollection 2024 Jul.
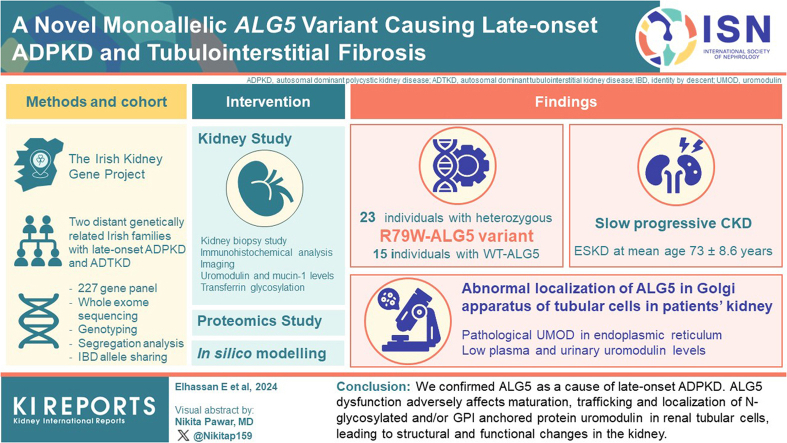
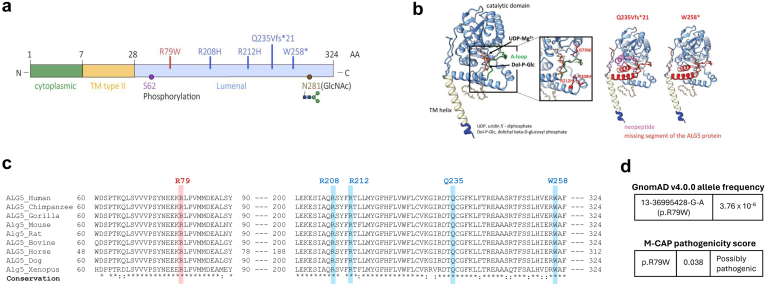
Monoallelic variants in the gene encoding asparagine-linked glycosylation protein 5 homolog (ALG5) have been recently shown to disrupt polycystin-1 (PC1) maturation and trafficking via underglycosylation, causing an autosomal dominant polycystic kidney disease-like (ADPKD-like) phenotype and interstitial fibrosis. In this report, we present clinical, genetic, histopathologic, and protein structure and functional correlates of a new ALG5 variant, p.R79W, that we identified in 2 distant genetically related Irish families displaying an atypical late-onset ADPKD phenotype combined with tubulointerstitial damage.
Whole exome and targeted sequencing were used for segregation analysis of available relatives. This was followed by immunohistochemistry examinations of kidney biopsies, and targeted (UMOD, MUC1) and untargeted plasma proteome and N-glycomic studies.
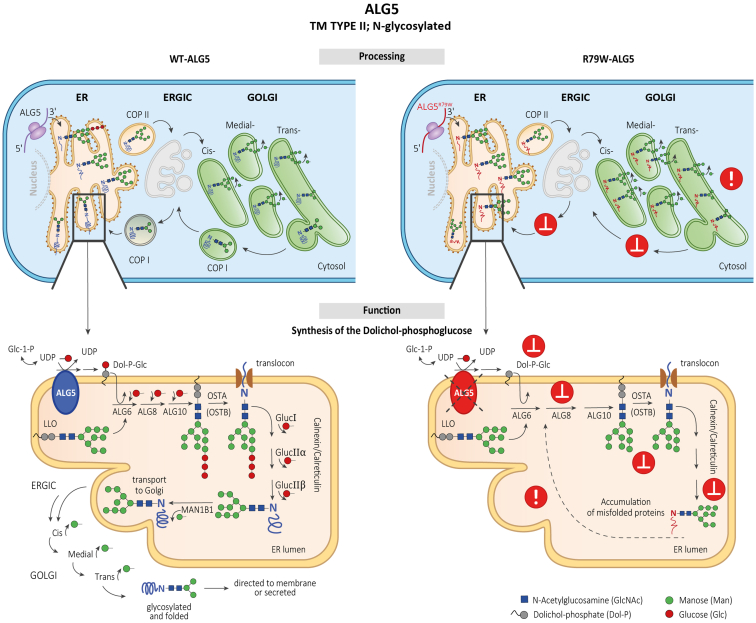
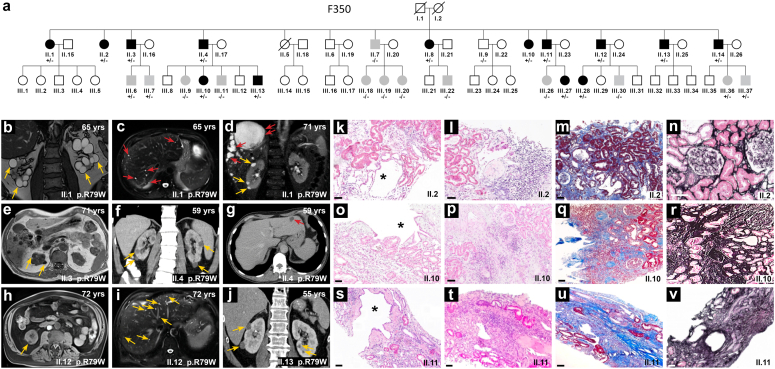
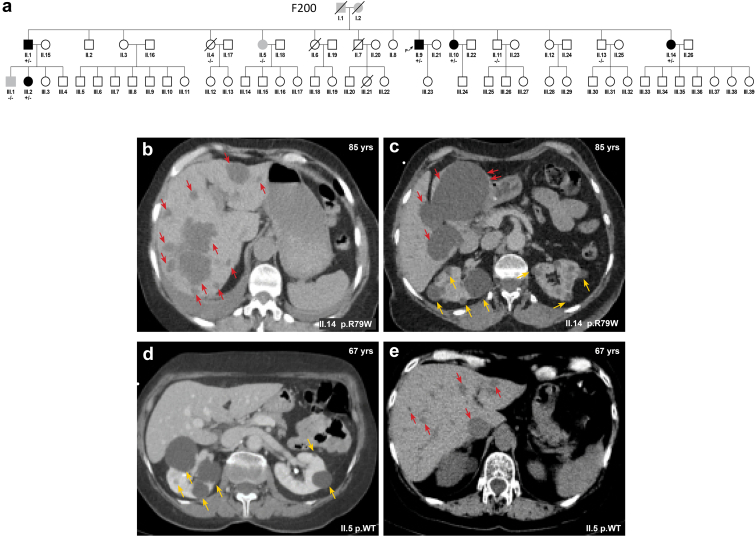
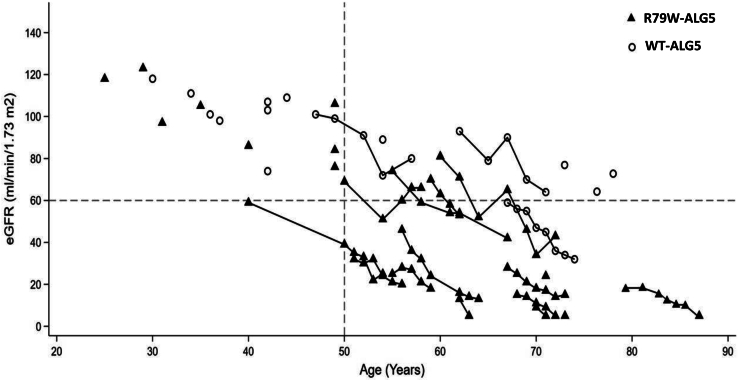
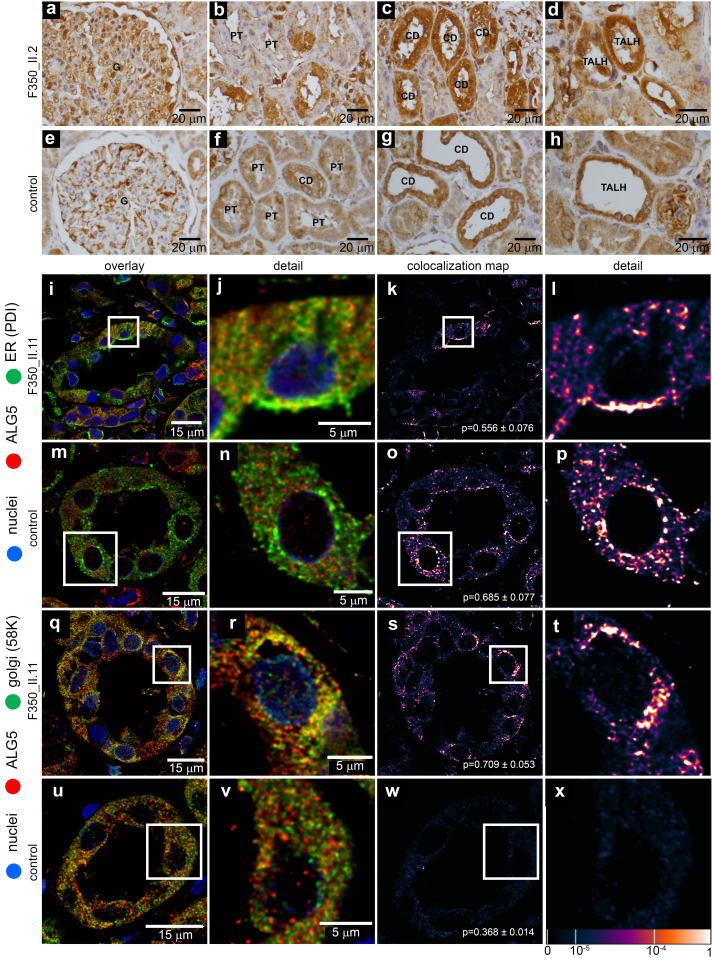
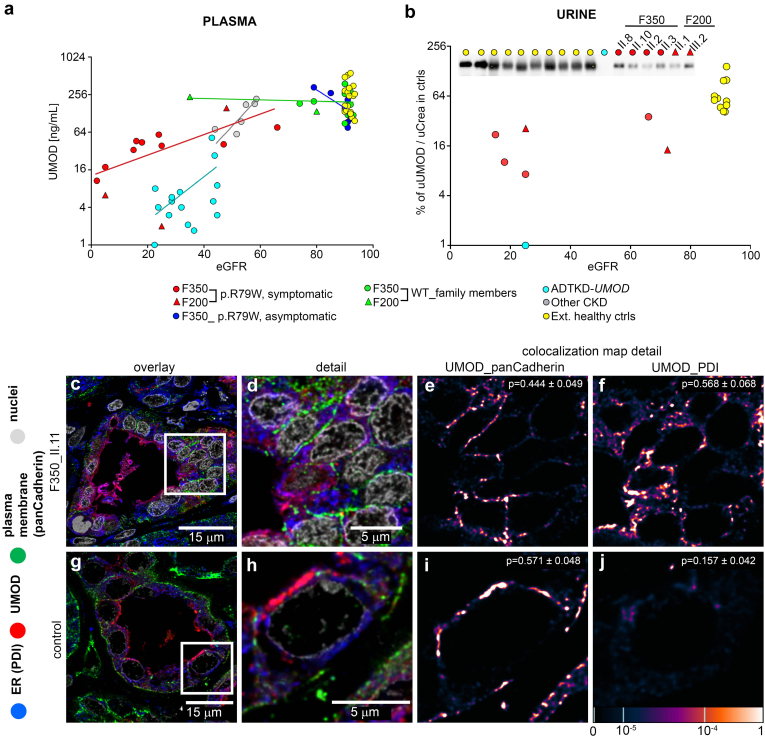
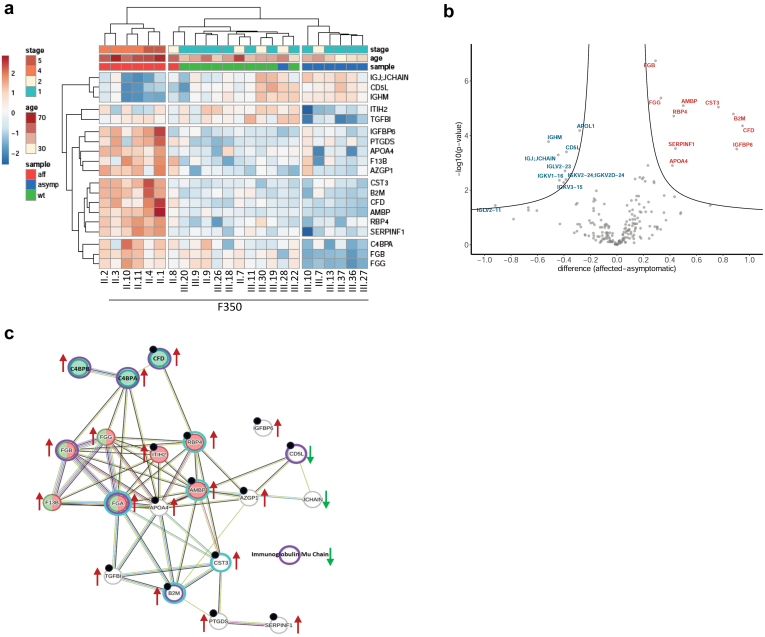
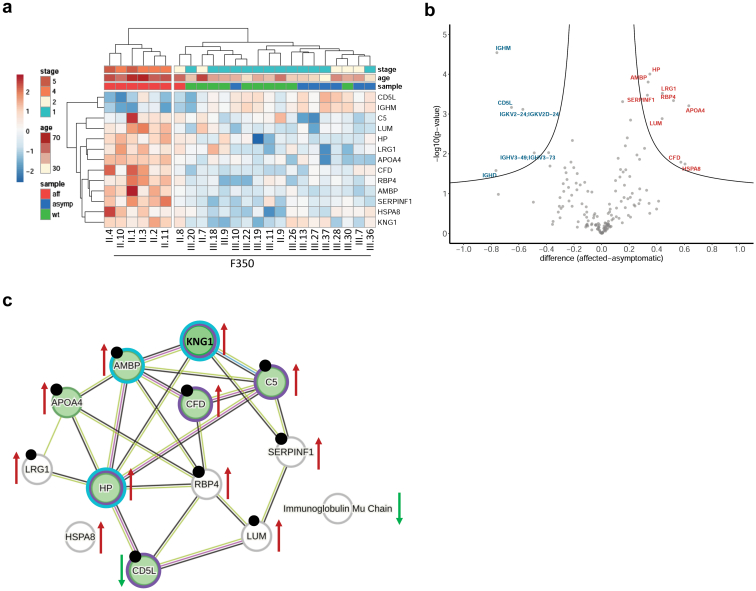
We identified a monoallelic variant [GRCh37 (NM_013338.5): g.37569565G>A, c.235C>T; p.R79W] that cosegregates in 23 individuals, of whom 18 were clinically affected. We detected abnormal localization of ALG5 in the Golgi apparatus of renal tubular cells in patients' kidney specimens. Further, we detected the pathological accumulation of uromodulin, an N-glycosylated glycosylphosphatidylinositol (GPI)-anchored protein, in the endoplasmic reticulum (ER), but not mucin-1, an O- and N-glycosylated protein. Biochemical investigation revealed decreased plasma and urinary uromodulin levels in clinically affected individuals. Proteomic and glycoproteomic profiling revealed the dysregulation of chronic kidney disease (CKD)-associated proteins.
ALG5 dysfunction adversely affects maturation and trafficking of N-glycosylated and GPI anchored protein uromodulin, leading to structural and functional changes in the kidney. Our findings confirm ALG5 as a cause of late-onset ADPKD and provide additional insight into the molecular mechanisms of ADPKD-.
最近研究表明,编码天冬酰胺连接糖基化蛋白5同源物(ALG5)的基因中的单等位基因变异通过糖基化不足破坏多囊蛋白-1(PC1)的成熟和运输,导致常染色体显性多囊肾病样(ADPKD样)表型和间质纤维化。在本报告中,我们展示了一个新的ALG5变异体p.R79W的临床、遗传、组织病理学、蛋白质结构和功能相关性,该变异体是我们在两个有远亲关系的爱尔兰家族中发现的,这些家族表现出非典型的迟发性ADPKD表型并伴有肾小管间质损伤。
采用全外显子测序和靶向测序对现有亲属进行分离分析。随后对肾活检组织进行免疫组化检查,并进行靶向(UMOD、MUC1)和非靶向血浆蛋白质组及N-糖组学研究。
我们鉴定出一个单等位基因变异体[GRCh37(NM_013338.5):g.37569565G>A,c.235C>T;p.R79W],该变异体在23名个体中共同分离,其中18名个体有临床症状。我们在患者肾脏标本的肾小管细胞高尔基体中检测到ALG5的异常定位。此外,我们在内质网(ER)中检测到尿调节蛋白(一种N-糖基化的糖基磷脂酰肌醇(GPI)锚定蛋白)的病理性积累,但未检测到粘蛋白-1(一种O-和N-糖基化蛋白)的积累。生化研究显示,有临床症状的个体血浆和尿液中的尿调节蛋白水平降低。蛋白质组学和糖蛋白质组学分析揭示了慢性肾脏病(CKD)相关蛋白的失调。
ALG5功能障碍对N-糖基化和GPI锚定蛋白尿调节蛋白的成熟和运输产生不利影响,导致肾脏结构和功能发生变化。我们的研究结果证实ALG5是迟发性ADPKD的一个病因,并为ADPKD的分子机制提供了更多见解。