Fonseca Emanuel M, Tran Linh N, Mendoza Hannah, Gutenkunst Ryan N
Department of Molecular and Cellular Biology, University of Arizona, Tucson, AZ 85721, USA.
bioRxiv. 2024 Jul 23:2024.07.19.604366. doi: 10.1101/2024.07.19.604366.
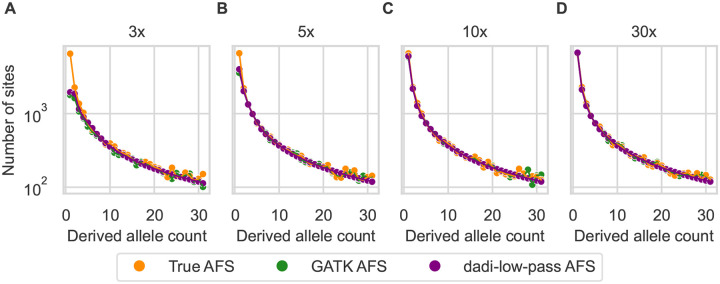
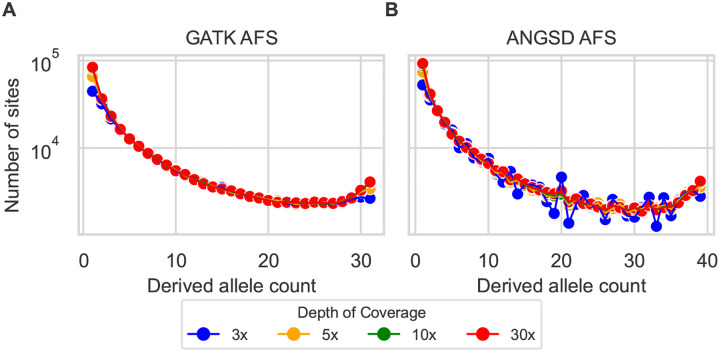
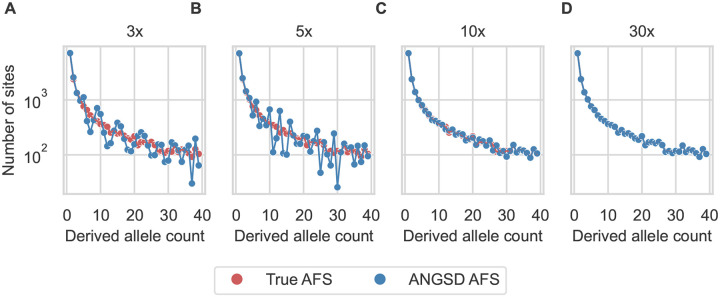
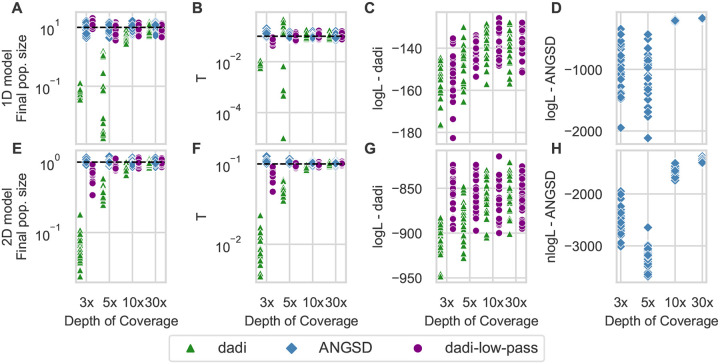
Low-pass genome sequencing is cost-effective and enables analysis of large cohorts. However, it introduces biases by reducing heterozygous genotypes and low-frequency alleles, impacting subsequent analyses such as demographic history inference. We developed a probabilistic model of low-pass biases from the Genome Analysis Toolkit (GATK) multi-sample calling pipeline, and we implemented it in the population genomic inference software dadi. We evaluated the model using simulated low-pass datasets and found that it alleviated low-pass biases in inferred demographic parameters. We further validated the model by downsampling 1000 Genomes Project data, demonstrating its effectiveness on real data. Our model is widely applicable and substantially improves model-based inferences from low-pass population genomic data.
低覆盖度基因组测序具有成本效益,能够对大规模队列进行分析。然而,它通过减少杂合基因型和低频等位基因引入偏差,影响后续分析,如人口历史推断。我们从基因组分析工具包(GATK)多样本调用流程中开发了一个低覆盖度偏差的概率模型,并将其应用于群体基因组推断软件dadi中。我们使用模拟的低覆盖度数据集评估了该模型,发现它减轻了推断人口参数中的低覆盖度偏差。我们通过对千人基因组计划数据进行下采样进一步验证了该模型,证明了其在真实数据上的有效性。我们的模型具有广泛的适用性,显著改进了基于低覆盖度群体基因组数据的模型推断。