Internal Medicine, Reference Center for Lysosomal Diseases (CRML), GH Diaconesses Croix Saint-Simon, Paris, France.
Reference Center for Inherited Metabolic Diseases, Hospices Civils de Lyon, Bron, France.
Orphanet J Rare Dis. 2024 Aug 5;19(1):289. doi: 10.1186/s13023-024-03234-6.
Acid sphingomyelinase deficiency (ASMD) or Niemann-Pick disease types A, A/B, and B is a progressive, life-limiting, autosomal recessive disorder caused by sphingomyelin phosphodiesterase 1 (SMPD1) gene mutations. There is a need to increase the understanding of morbidity and mortality across children to adults diagnosed with ASMD.
This observational retrospective survey analysed medical records of patients with ASMD with retrievable data from 27 hospitals in France, diagnosed/followed up between 1 January 1990 and 31 December 2020. Eligible records were abstracted to collect demographic, medical/developmental history, and mortality data. Survival outcomes were estimated from birth until death using Kaplan-Meier survival analyses; standardised mortality ratio (SMR) was also explored.
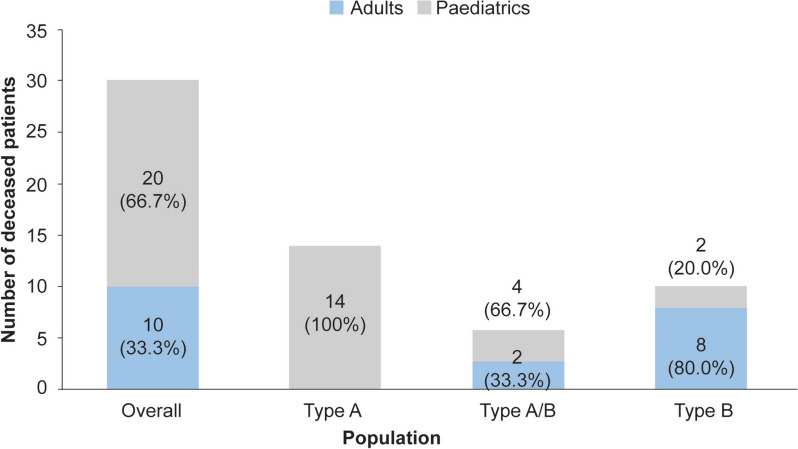
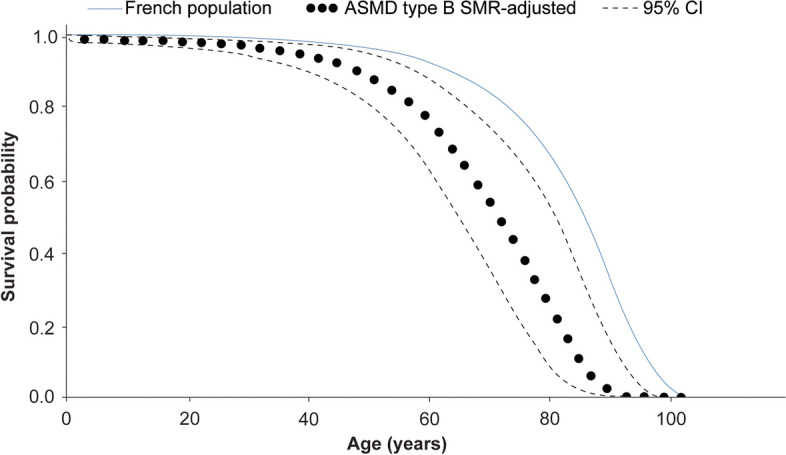
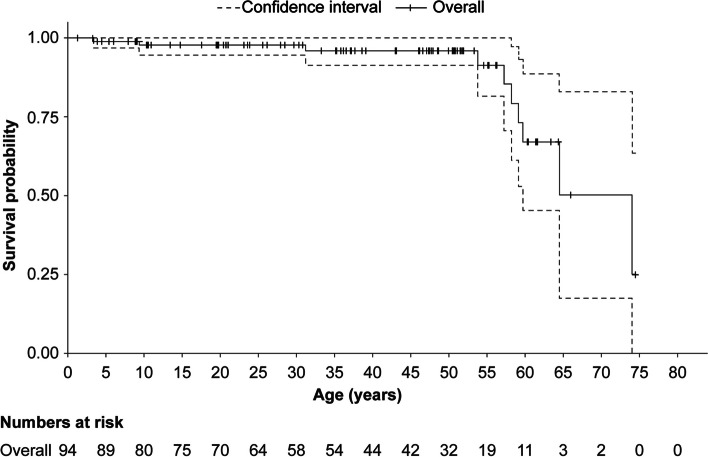
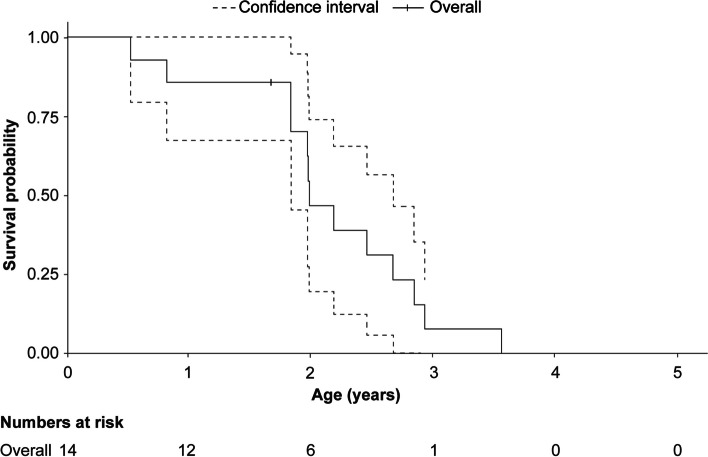
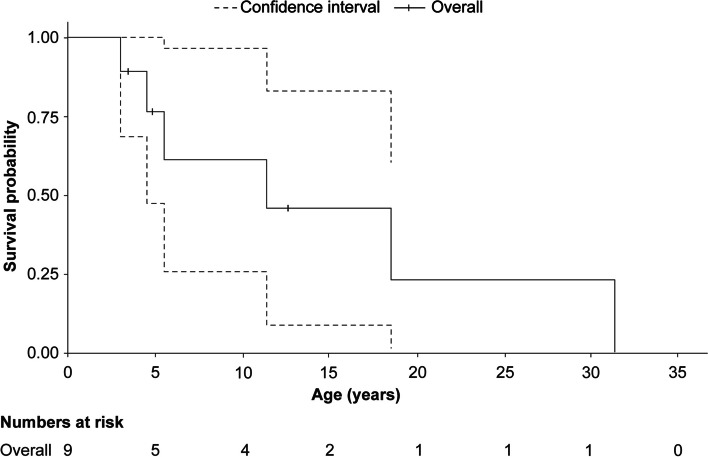
A total of 118 medical records of patients with ASMD (type B [n = 94], type A [n = 15], and type A/B [n = 9]) were assessed. The majority of patients were males (63.6%); the median [range] age at diagnosis was 8.0 [1.0-18.0] months (type A), 1.0 [0-3] year (type A/B), and 5.5 [0-73] years (type B). Overall, 30 patients were deceased at the study completion date; the median [range] age at death for patients with ASMD type A (n = 14) was 1 [0-3.6] year, type A/B (n = 6) was 8.5 [3.0-30.9] years, and type B (n = 10) was 57.6 [3.4-74.1] years. The median [95% confidence interval (CI)] survival age from birth in patients with ASMD type A and type A/B was 2.0 [1.8-2.7] years and 11.4 [5.5-18.5] years, respectively. Survival analysis in ASMD type B was explored using SMR [95% CI] analysis (3.5 [1.6-5.9]), which showed that age-specific deaths in the ASMD type B population were 3.5 times more frequent than those in the general French population. The causes of death were mostly severe progressive neurodegeneration (type A: 16.7%), cancer (type B: 16.7%), or unspecified (across groups: 33.3%).
This study illustrated a substantial burden of illness with high mortality rates in patients with ASMD, including adults with ASMD type B, in France.
酸性鞘磷脂酶缺乏症(ASMD)或尼曼-皮克病 A、A/B 和 B 型是一种进行性、危及生命的常染色体隐性遗传病,由鞘磷脂磷酸二酯酶 1(SMPD1)基因突变引起。需要提高对从儿童到成年确诊为 ASMD 患者发病率和死亡率的认识。
本观察性回顾性调查分析了法国 27 家医院的 ASMD 患者的病历,这些患者的诊断/随访时间为 1990 年 1 月 1 日至 2020 年 12 月 31 日。提取符合条件的记录,以收集人口统计学、医疗/发育史和死亡率数据。使用 Kaplan-Meier 生存分析估计从出生到死亡的生存结果;还探讨了标准化死亡率比(SMR)。
共评估了 118 例 ASMD 患者(B 型[94 例]、A 型[15 例]和 A/B 型[9 例])的病历。大多数患者为男性(63.6%);诊断时的中位[范围]年龄为 8.0[1.0-18.0]个月(A 型)、1.0[0-3]岁(A/B 型)和 5.5[0-73]岁(B 型)。研究完成时,共有 30 名患者死亡;A 型(n=14)、A/B 型(n=6)和 B 型(n=10)患者的中位[范围]死亡年龄分别为 1[0-3.6]岁、8.5[3.0-30.9]岁和 57.6[3.4-74.1]岁。ASMD A 型和 A/B 型患者的中位[95%置信区间(CI)]出生后生存年龄分别为 2.0[1.8-2.7]年和 11.4[5.5-18.5]年。B 型 ASMD 的生存分析使用 SMR[95%CI]分析(3.5[1.6-5.9])进行了探索,表明 B 型 ASMD 人群的特定年龄死亡比法国一般人群高 3.5 倍。死亡原因主要为严重进行性神经退行性变(A 型:16.7%)、癌症(B 型:16.7%)或不明原因(各组:33.3%)。
本研究表明,法国 ASMD 患者,包括 B 型成年患者,疾病负担严重,死亡率高。