Jiang Ling, Zhao Yu, Liu Fang, Huang Yun, Zhang Yujiao, Yuan Baoyi, Cheng Jiaying, Yan Ping, Ni Jinle, Jiang Yongshuai, Wu Quan, Jiang Xuejie
Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou, China.
Department of Hematology, The Third Affiliated Hospital of Southern Medical University, Guangzhou, China.
Cell Commun Signal. 2024 Aug 7;22(1):391. doi: 10.1186/s12964-024-01774-9.
Approximately 25-30% of patients with acute myeloid leukemia (AML) have FMS-like receptor tyrosine kinase-3 (FLT3) mutations that contribute to disease progression and poor prognosis. Prolonged exposure to FLT3 tyrosine kinase inhibitors (TKIs) often results in limited clinical responses due to diverse compensatory survival signals. Therefore, there is an urgent need to elucidate the mechanisms underlying FLT3 TKI resistance. Dysregulated sphingolipid metabolism frequently contributes to cancer progression and a poor therapeutic response. However, its relationship with TKI sensitivity in FLT3-mutated AML remains unknown. Thus, we aimed to assess mechanisms of FLT3 TKI resistance in AML.
We performed lipidomics profiling, RNA-seq, qRT-PCR, and enzyme-linked immunosorbent assays to determine potential drivers of sorafenib resistance. FLT3 signaling was inhibited by sorafenib or quizartinib, and SPHK1 was inhibited by using an antagonist or via knockdown. Cell growth and apoptosis were assessed in FLT3-mutated and wild-type AML cell lines via Cell counting kit-8, PI staining, and Annexin-V/7AAD assays. Western blotting and immunofluorescence assays were employed to explore the underlying molecular mechanisms through rescue experiments using SPHK1 overexpression and exogenous S1P, as well as inhibitors of S1P2, β-catenin, PP2A, and GSK3β. Xenograft murine model, patient samples, and publicly available data were analyzed to corroborate our in vitro results.
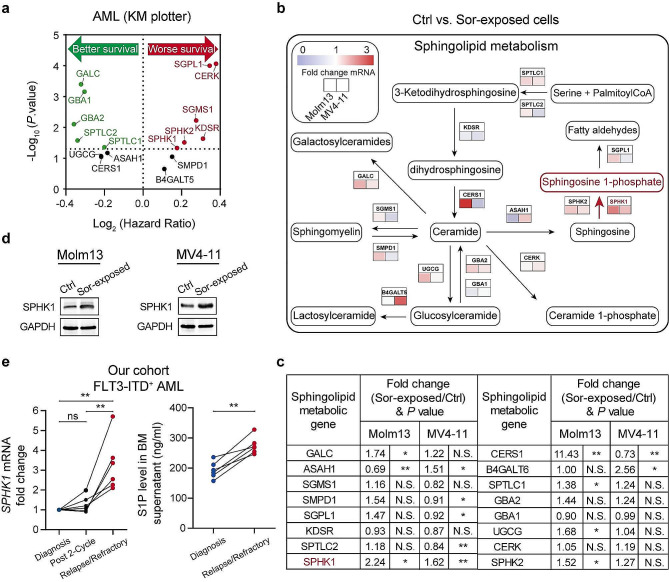
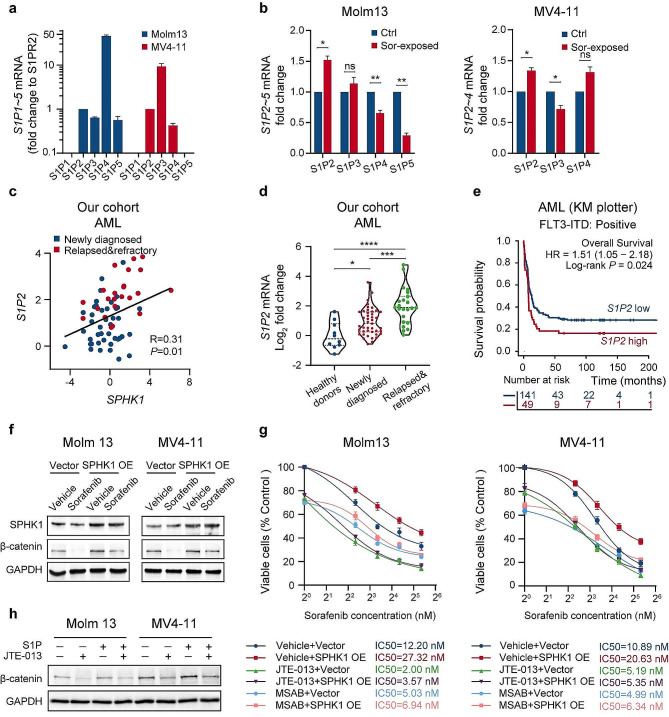
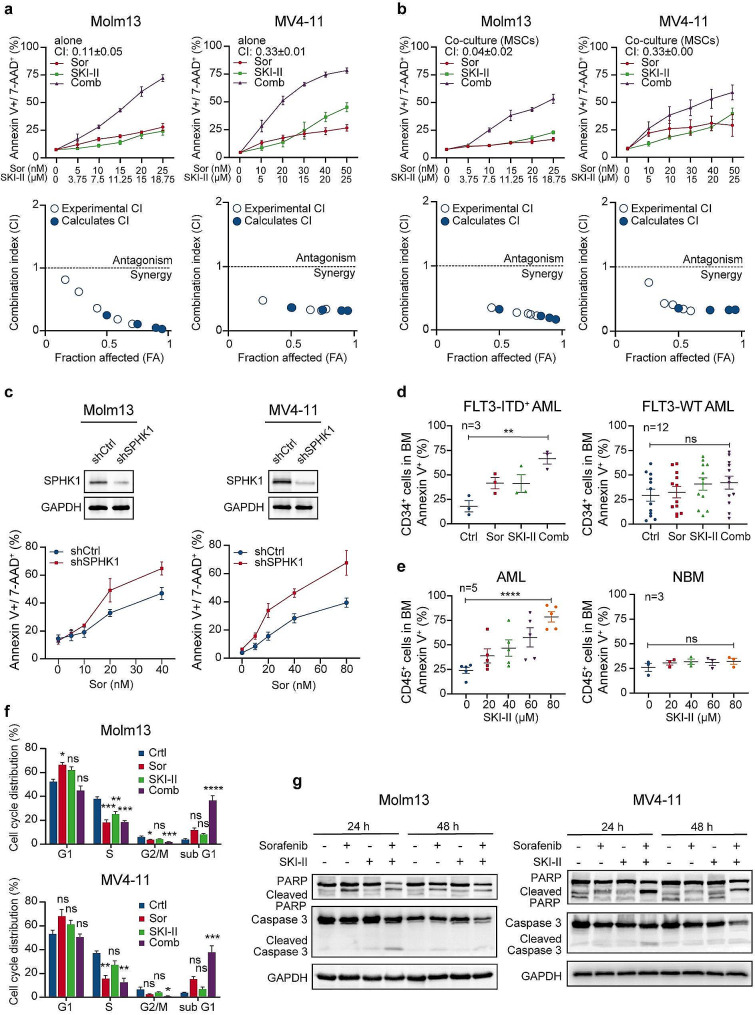
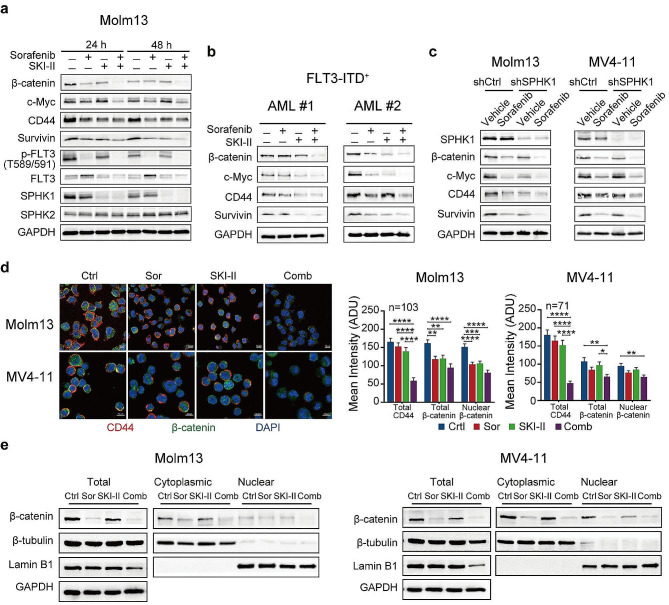
We demonstrate that long-term sorafenib treatment upregulates SPHK1/sphingosine-1-phosphate (S1P) signaling, which in turn positively modulates β-catenin signaling to counteract TKI-mediated suppression of FLT3-mutated AML cells via the S1P2 receptor. Genetic or pharmacological inhibition of SPHK1 potently enhanced the TKI-mediated inhibition of proliferation and apoptosis induction in FLT3-mutated AML cells in vitro. SPHK1 knockdown enhanced sorafenib efficacy and improved survival of AML-xenografted mice. Mechanistically, targeting the SPHK1/S1P/S1P2 signaling synergizes with FLT3 TKIs to inhibit β-catenin activity by activating the protein phosphatase 2 A (PP2A)-glycogen synthase kinase 3β (GSK3β) pathway.
These findings establish the sphingolipid metabolic enzyme SPHK1 as a regulator of TKI sensitivity and suggest that combining SPHK1 inhibition with TKIs could be an effective approach for treating FLT3-mutated AML.
大约25%-30%的急性髓系白血病(AML)患者存在FMS样受体酪氨酸激酶-3(FLT3)突变,这些突变会导致疾病进展和预后不良。由于存在多种代偿性生存信号,长期暴露于FLT3酪氨酸激酶抑制剂(TKIs)通常会导致临床反应有限。因此,迫切需要阐明FLT3 TKI耐药的潜在机制。鞘脂代谢失调常常促进癌症进展并导致治疗反应不佳。然而,其与FLT3突变型AML中TKI敏感性的关系尚不清楚。因此,我们旨在评估AML中FLT3 TKI耐药的机制。
我们进行了脂质组学分析、RNA测序、qRT-PCR和酶联免疫吸附测定,以确定索拉非尼耐药的潜在驱动因素。索拉非尼或奎扎替尼抑制FLT3信号传导,使用拮抗剂或通过敲低抑制SPHK1。通过细胞计数试剂盒-8、PI染色和Annexin-V/7AAD测定评估FLT3突变型和野生型AML细胞系中的细胞生长和凋亡。采用蛋白质免疫印迹法和免疫荧光测定法,通过使用SPHK1过表达和外源性S1P以及S1P2、β-连环蛋白、PP2A和GSK3β抑制剂的挽救实验来探索潜在的分子机制。分析异种移植小鼠模型、患者样本和公开可用数据以证实我们的体外实验结果。
我们证明,长期索拉非尼治疗会上调SPHK1/鞘氨醇-1-磷酸(S1P)信号传导,进而正向调节β-连环蛋白信号传导,以通过S1P2受体抵消TKI介导的对FLT3突变型AML细胞的抑制作用。对SPHK1进行基因或药理学抑制可有效增强TKI介导的对体外FLT3突变型AML细胞增殖的抑制作用和凋亡诱导作用。敲低SPHK1可增强索拉非尼疗效并提高AML异种移植小鼠的存活率。从机制上讲,靶向SPHK1/S1P/S1P2信号传导与FLT3 TKIs协同作用,通过激活蛋白磷酸酶2A(PP2A)-糖原合酶激酶3β(GSK3β)途径抑制β-连环蛋白活性。
这些发现确立了鞘脂代谢酶SPHK1作为TKI敏感性的调节因子,并表明将SPHK1抑制与TKIs联合使用可能是治疗FLT3突变型AML的有效方法。