Aßmann Eva, Agrawal Shelesh, Orschler Laura, Böttcher Sindy, Lackner Susanne, Hölzer Martin
Genome Competence Center (MF1), Robert Koch Institute, Berlin 13353, Germany.
Center for Artificial Intelligence in Public Health Research (ZKI-PH), Robert Koch Institute, Berlin 13353, Germany.
Gigascience. 2024 Jan 2;13. doi: 10.1093/gigascience/giae051.
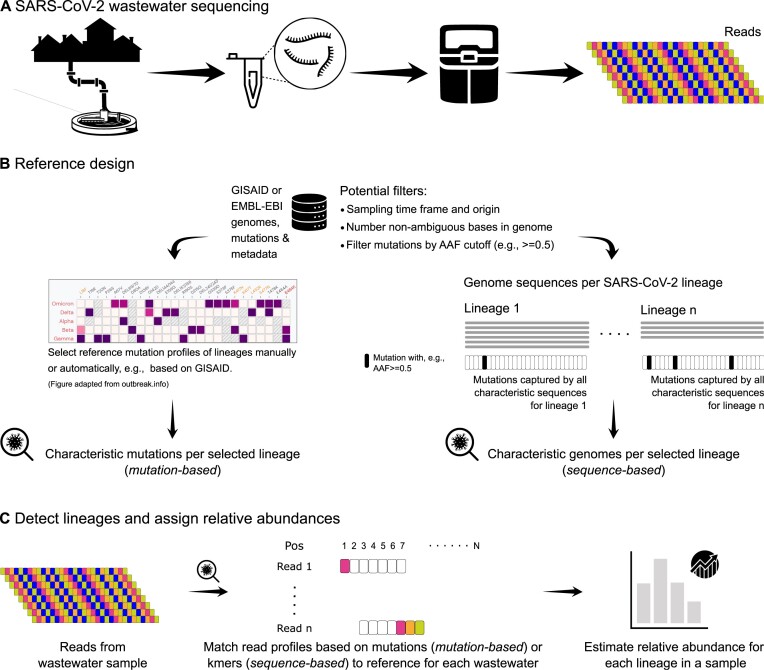
Sequencing of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA from wastewater samples has emerged as a valuable tool for detecting the presence and relative abundances of SARS-CoV-2 variants in a community. By analyzing the viral genetic material present in wastewater, researchers and public health authorities can gain early insights into the spread of virus lineages and emerging mutations. Constructing reference datasets from known SARS-CoV-2 lineages and their mutation profiles has become state-of-the-art for assigning viral lineages and their relative abundances from wastewater sequencing data. However, selecting reference sequences or mutations directly affects the predictive power.
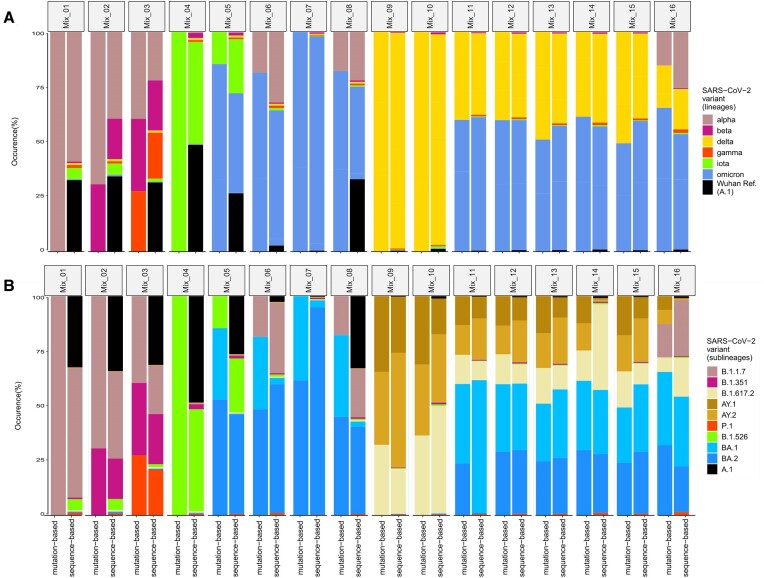
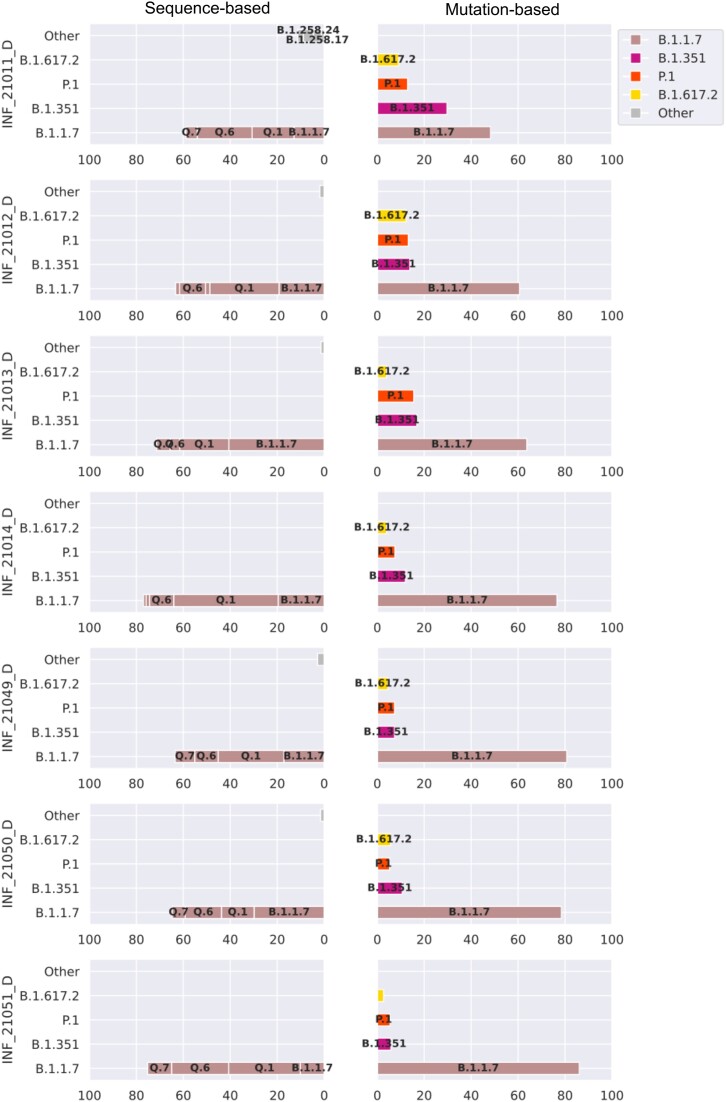
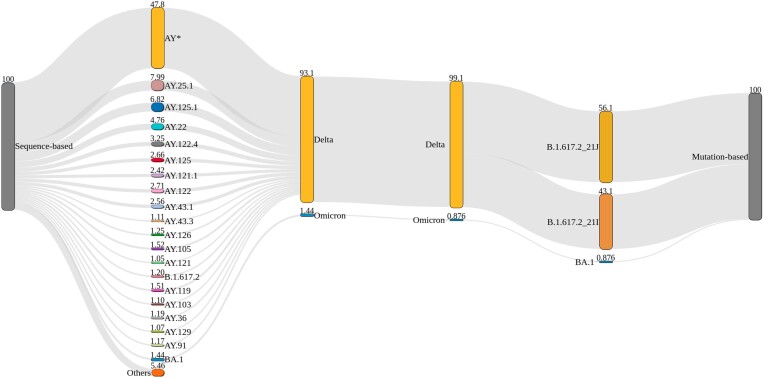
Here, we show the impact of a mutation- and sequence-based reference reconstruction for SARS-CoV-2 abundance estimation. We benchmark 3 datasets: (i) synthetic "spike-in"' mixtures; (ii) German wastewater samples from early 2021, mainly comprising Alpha; and (iii) samples obtained from wastewater at an international airport in Germany from the end of 2021, including first signals of Omicron. The 2 approaches differ in sublineage detection, with the marker mutation-based method, in particular, being challenged by the increasing number of mutations and lineages. However, the estimations of both approaches depend on selecting representative references and optimized parameter settings. By performing parameter escalation experiments, we demonstrate the effects of reference size and alternative allele frequency cutoffs for abundance estimation. We show how different parameter settings can lead to different results for our test datasets and illustrate the effects of virus lineage composition of wastewater samples and references.
Our study highlights current computational challenges, focusing on the general reference design, which directly impacts abundance allocations. We illustrate advantages and disadvantages that may be relevant for further developments in the wastewater community and in the context of defining robust quality metrics.
对污水样本中的严重急性呼吸综合征冠状病毒2(SARS-CoV-2)RNA进行测序已成为检测社区中SARS-CoV-2变体的存在及其相对丰度的重要工具。通过分析污水中存在的病毒遗传物质,研究人员和公共卫生当局可以对病毒谱系的传播和新出现的突变有早期了解。根据已知的SARS-CoV-2谱系及其突变谱构建参考数据集已成为从污水测序数据中确定病毒谱系及其相对丰度的最新技术。然而,直接选择参考序列或突变会影响预测能力。
在这里,我们展示了基于突变和序列的参考重建对SARS-CoV-2丰度估计的影响。我们对3个数据集进行了基准测试:(i)合成的“加标”混合物;(ii)2021年初的德国污水样本,主要包含阿尔法变体;(iii)2021年底从德国一个国际机场的污水中获取的样本,包括奥密克戎的首个信号。这两种方法在亚谱系检测方面存在差异,特别是基于标记突变的方法受到突变和谱系数量增加的挑战。然而,两种方法的估计都取决于选择代表性参考和优化参数设置。通过进行参数递增实验,我们展示了参考大小和替代等位基因频率截止值对丰度估计的影响。我们展示了不同的参数设置如何导致我们测试数据集的不同结果,并说明了污水样本和参考的病毒谱系组成的影响。
我们的研究突出了当前的计算挑战,重点是直接影响丰度分配的通用参考设计。我们阐述了对于污水领域进一步发展以及在定义稳健质量指标背景下可能相关的优点和缺点。