School of Basic Medical Sciences and Intelligent Medicine Institute, Fudan University, Shanghai, 200032, China.
Shanghai Institute of Stem Cell Research and Clinical Translation, Shanghai, 200120, China.
J Transl Med. 2024 Aug 12;22(1):756. doi: 10.1186/s12967-024-05567-z.
Decoding human genomic sequences requires comprehensive analysis of DNA sequence functionality. Through computational and experimental approaches, researchers have studied the genotype-phenotype relationship and generate important datasets that help unravel complicated genetic blueprints. Thus, the recently developed artificial intelligence methods can be used to interpret the functions of those DNA sequences.
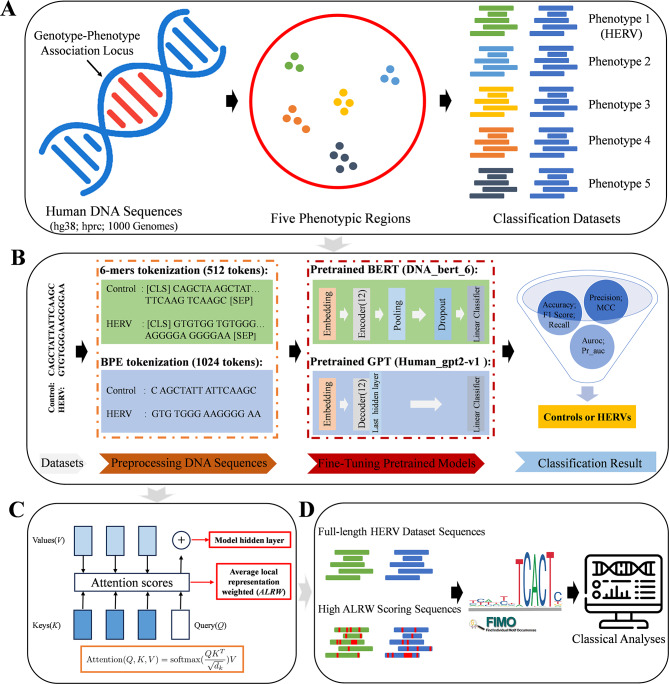
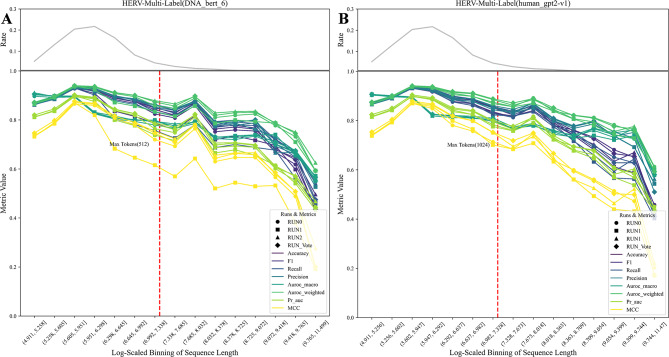
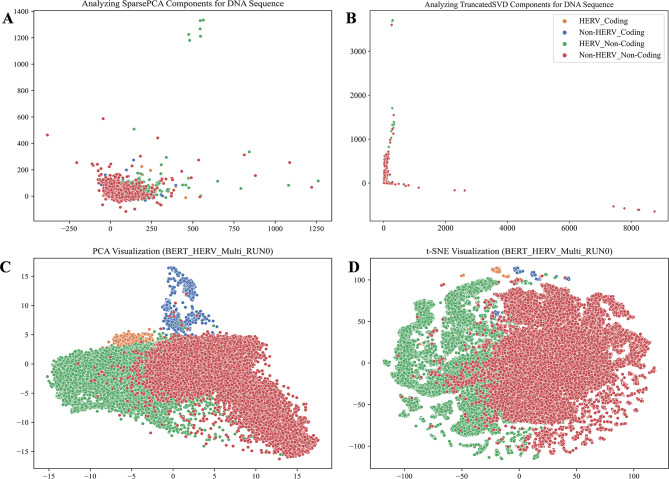
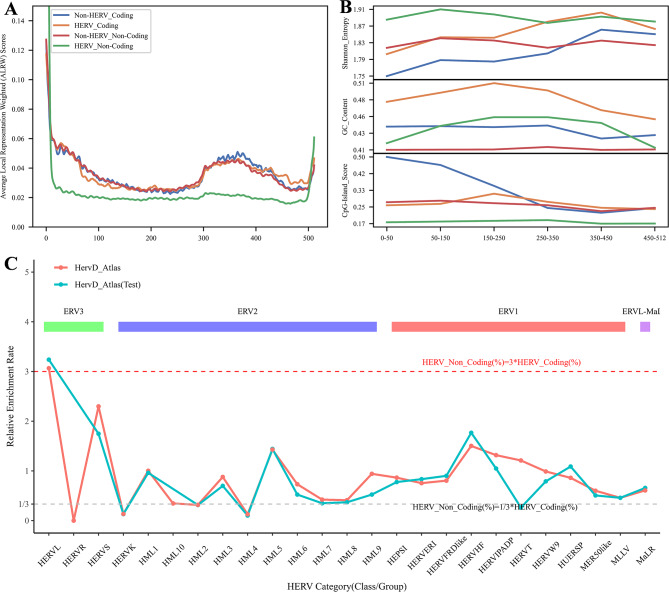
This study explores the use of deep learning, particularly pre-trained genomic models like DNA_bert_6 and human_gpt2-v1, in interpreting and representing human genome sequences. Initially, we meticulously constructed multiple datasets linking genotypes and phenotypes to fine-tune those models for precise DNA sequence classification. Additionally, we evaluate the influence of sequence length on classification results and analyze the impact of feature extraction in the hidden layers of our model using the HERV dataset. To enhance our understanding of phenotype-specific patterns recognized by the model, we perform enrichment, pathogenicity and conservation analyzes of specific motifs in the human endogenous retrovirus (HERV) sequence with high average local representation weight (ALRW) scores.
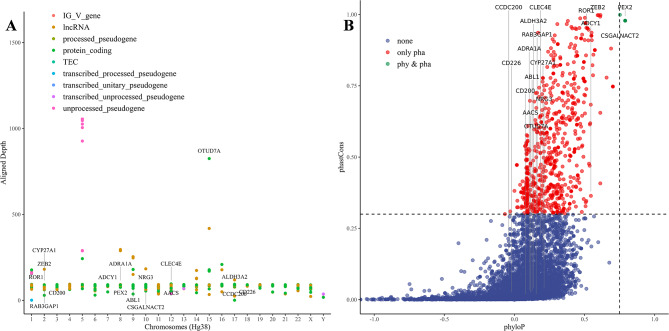
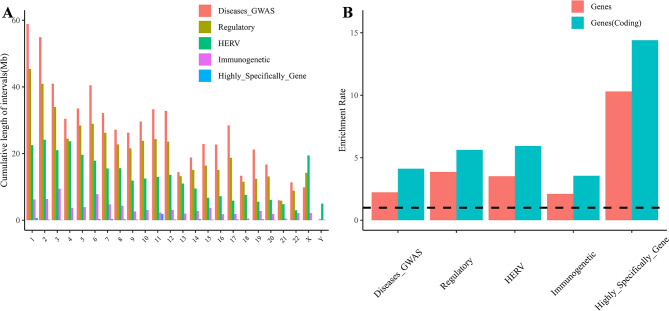
We have constructed multiple genotype-phenotype datasets displaying commendable classification performance in comparison with random genomic sequences, particularly in the HERV dataset, which achieved binary and multi-classification accuracies and F1 values exceeding 0.935 and 0.888, respectively. Notably, the fine-tuning of the HERV dataset not only improved our ability to identify and distinguish diverse information types within DNA sequences but also successfully identified specific motifs associated with neurological disorders and cancers in regions with high ALRW scores. Subsequent analysis of these motifs shed light on the adaptive responses of species to environmental pressures and their co-evolution with pathogens.
These findings highlight the potential of pre-trained genomic models in learning DNA sequence representations, particularly when utilizing the HERV dataset, and provide valuable insights for future research endeavors. This study represents an innovative strategy that combines pre-trained genomic model representations with classical methods for analyzing the functionality of genome sequences, thereby promoting cross-fertilization between genomics and artificial intelligence.
解码人类基因组序列需要全面分析 DNA 序列的功能。通过计算和实验方法,研究人员研究了基因型-表型关系,并生成了重要的数据集,帮助揭示复杂的遗传蓝图。因此,最近开发的人工智能方法可以用于解释这些 DNA 序列的功能。
本研究探讨了深度学习,特别是预训练的基因组模型,如 DNA_bert_6 和 human_gpt2-v1,在解释和表示人类基因组序列中的应用。我们首先精心构建了多个将基因型和表型联系起来的数据集,以微调这些模型,从而实现精确的 DNA 序列分类。此外,我们还评估了序列长度对分类结果的影响,并使用 HERV 数据集分析了模型隐藏层中特征提取的影响。为了增强我们对模型识别的表型特异性模式的理解,我们对具有高平均局部表示权重 (ALRW) 得分的人类内源性逆转录病毒 (HERV) 序列中的特定基序进行了富集、致病性和保守性分析。
我们构建了多个基因型-表型数据集,与随机基因组序列相比,这些数据集显示出令人称赞的分类性能,特别是在 HERV 数据集上,二进制和多类分类的准确率和 F1 值分别超过 0.935 和 0.888。值得注意的是,对 HERV 数据集的微调不仅提高了我们识别和区分 DNA 序列中不同信息类型的能力,而且还成功地识别了与高 ALRW 得分区域中神经退行性疾病和癌症相关的特定基序。对这些基序的后续分析揭示了物种对环境压力的适应性反应及其与病原体的共同进化。
这些发现强调了预训练基因组模型在学习 DNA 序列表示方面的潜力,特别是在使用 HERV 数据集时,为未来的研究工作提供了有价值的见解。本研究代表了一种将预训练基因组模型表示与分析基因组序列功能的经典方法相结合的创新策略,从而促进了基因组学和人工智能之间的交叉融合。