Wiesenfarth Maximilian, Forouhideh-Wiesenfarth Yalda, Elmas Zeynep, Parlak Özlem, Weiland Ulrike, Herrmann Christine, Schuster Joachim, Freischmidt Axel, Müller Kathrin, Siebert Reiner, Günther Kornelia, Fröhlich Elke, Knehr Antje, Simak Tatiana, Bachhuber Franziska, Regensburger Martin, Petri Susanne, Klopstock Thomas, Reilich Peter, Schöberl Florian, Schumann Peggy, Körtvélyessy Peter, Meyer Thomas, Ruf Wolfgang P, Witzel Simon, Tumani Hayrettin, Brenner David, Dorst Johannes, Ludolph Albert C
Department of Neurology, Ulm University, Oberer Eselsberg 45, 89081, Ulm, Germany.
German Centre for Neurodegenerative Diseases (DZNE) Site Ulm, 89081, Ulm, Germany.
J Neurol. 2024 Oct;271(10):6667-6679. doi: 10.1007/s00415-024-12564-1. Epub 2024 Aug 14.
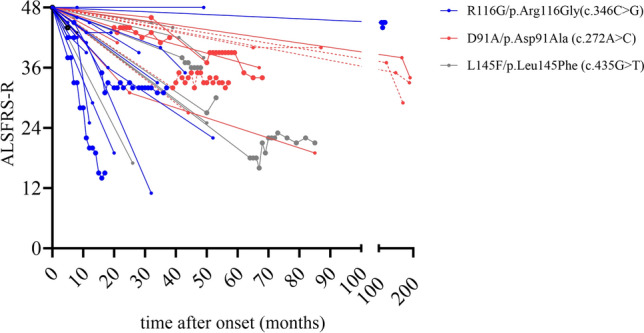
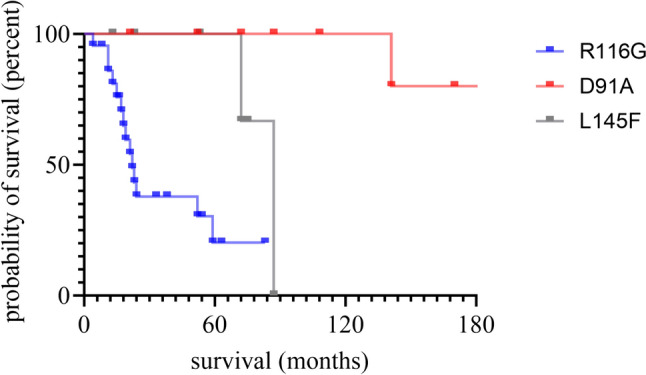
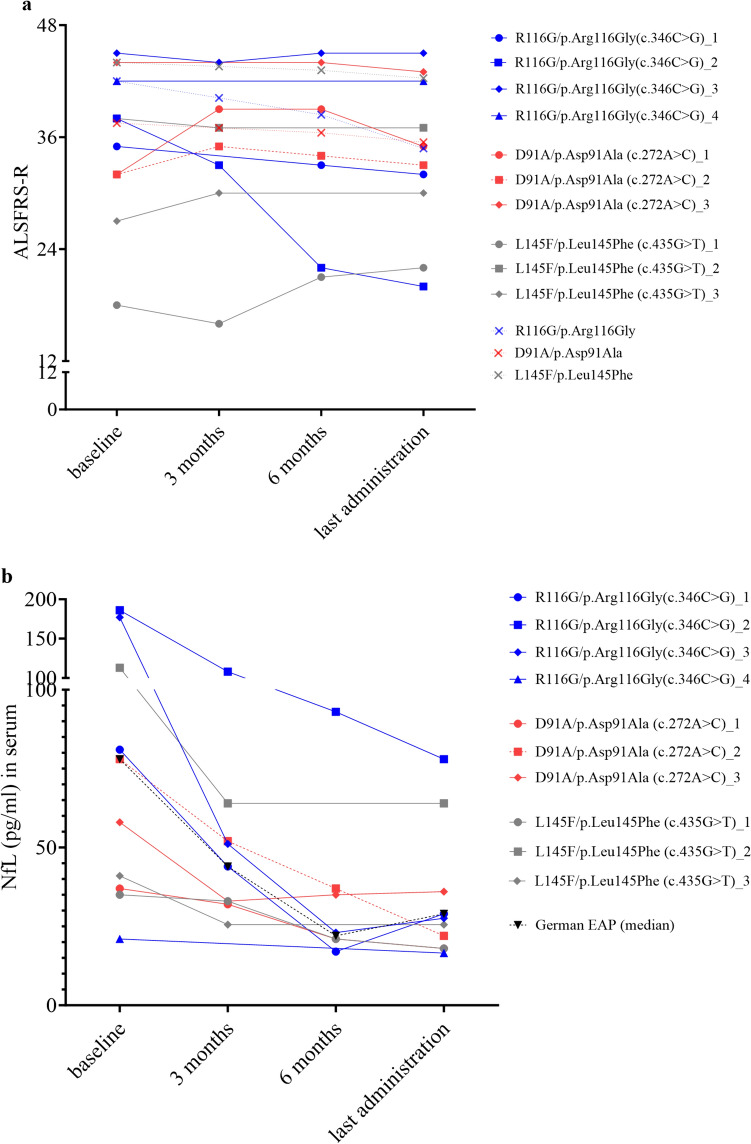
Pathogenic variants in the Cu/Zn superoxide dismutase (SOD1) gene can be detected in approximately 2% of sporadic and 11% of familial amyotrophic lateral sclerosis (ALS) patients in Europe. We analyzed the clinical phenotypes of 83 SOD1-ALS patients focusing on patients carrying the most frequent (likely) pathogenic variants (R116G, D91A, L145F) in Germany. Moreover, we describe the effect of tofersen treatment on ten patients carrying these variants. R116G patients showed the most aggressive course of disease with a median survival of 22.0 months compared to 198.0 months in D91A and 87.0 months in L145F patients (HR 7.71, 95% CI 2.89-20.58 vs. D91A; p < 0.001 and HR 4.25, 95% CI 1.55-11.67 vs. L145F; p = 0.02). Moreover, R116G patients had the fastest median ALSFRS-R progression rate with 0.12 (IQR 0.07-0.20) points lost per month. Median diagnostic delay was 10.0 months (IQR 5.5-11.5) and therefore shorter compared to 57.5 months (IQR 14.0-83.0) in D91A (p < 0.001) and 21.5 months (IQR 5.8-38.8) in L145F (p = 0.21) carriers. As opposed to D91A carriers (50.0%), 96.2% of R116G (p < 0.001) and 100.0% of L145F (p = 0.04) patients reported a positive family history. During tofersen treatment, all patients showed a reduction of neurofilament light chain (NfL) serum levels, independent of the SOD1 variant. Patients with SOD1-ALS carrying R116G, D91A, or L145F variants show commonalities, but also differences in their clinical phenotype, including a faster progression rate with shorter survival in R116G, and a comparatively benign disease course in D91A carriers.
在欧洲,约2%的散发性肌萎缩侧索硬化症(ALS)患者和11%的家族性ALS患者中可检测到铜/锌超氧化物歧化酶(SOD1)基因的致病变异。我们分析了83例SOD1-ALS患者的临床表型,重点关注德国携带最常见(可能)致病变异(R116G、D91A、L145F)的患者。此外,我们描述了托非生治疗对10例携带这些变异患者的影响。R116G患者的疾病进展最为迅速,中位生存期为22.0个月,而D91A患者为198.0个月,L145F患者为87.0个月(风险比7.71,95%置信区间2.89-20.58,与D91A相比;p<0.001;风险比4.25,95%置信区间1.55-11.67,与L145F相比;p=0.02)。此外,R116G患者的ALSFRS-R中位进展速度最快,每月下降0.12(四分位间距0.07-0.20)分。中位诊断延迟为10.0个月(四分位间距5.5-11.5),因此与D91A患者的57.5个月(四分位间距14.0-83.0)相比更短(p<0.001),与L145F携带者的21.5个月(四分位间距5.8-38.8)相比(p=0.21)。与D91A携带者(50.0%)不同,96.2%的R116G患者(p<0.001)和100.0%的L145F患者(p=0.04)报告有家族史阳性。在托非生治疗期间,所有患者的血清神经丝轻链(NfL)水平均降低,与SOD1变异无关。携带R116G、D91A或L145F变异的SOD1-ALS患者表现出共性,但临床表型也存在差异,包括R116G患者进展速度更快、生存期更短,而D91A携带者的疾病进程相对良性。