Institute of Computational Life Sciences, Zürich University of Applied Sciences (ZHAW), Wädenswil, Switzerland.
AI for Scientific Discovery, IBM Research Europe, Rüschlikon, Switzerland.
Front Immunol. 2024 Aug 5;15:1428773. doi: 10.3389/fimmu.2024.1428773. eCollection 2024.
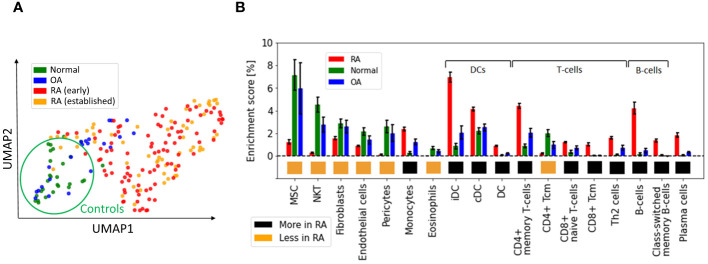
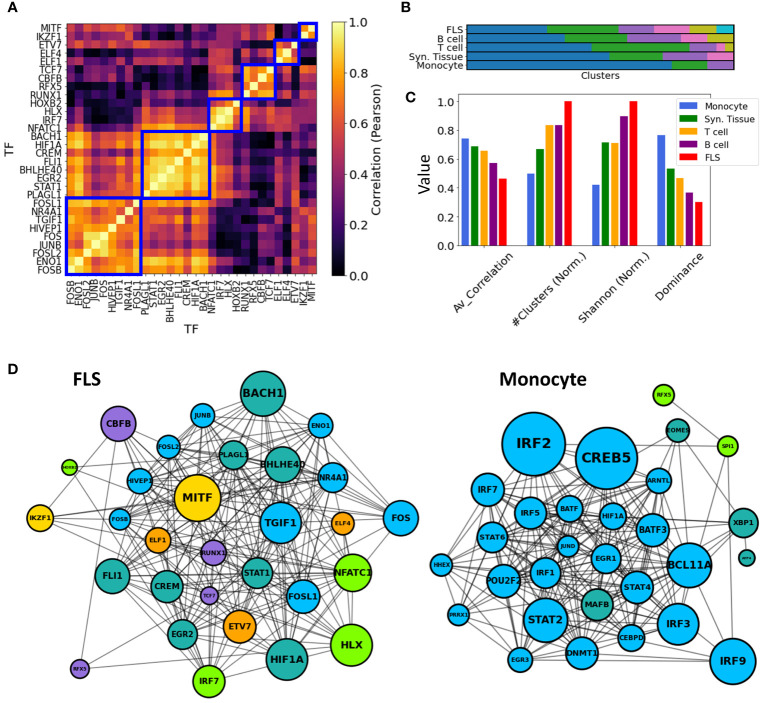
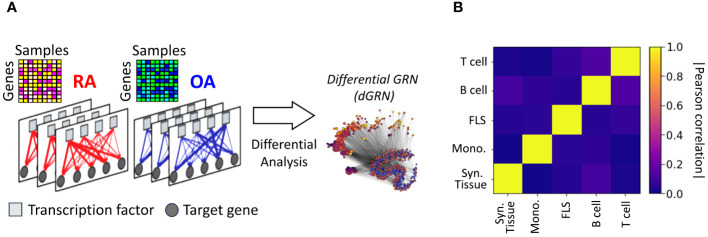
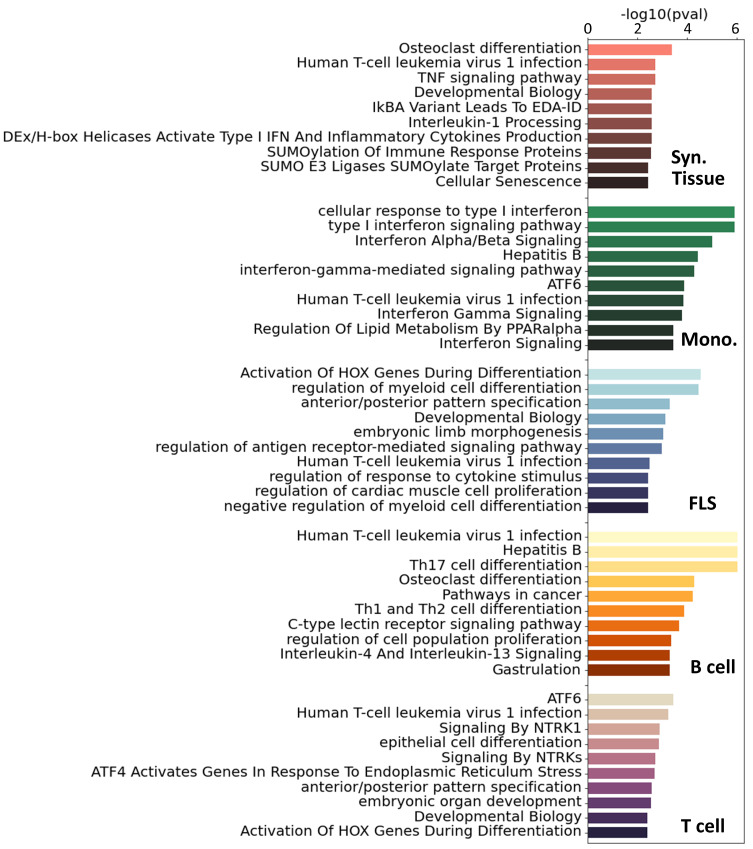
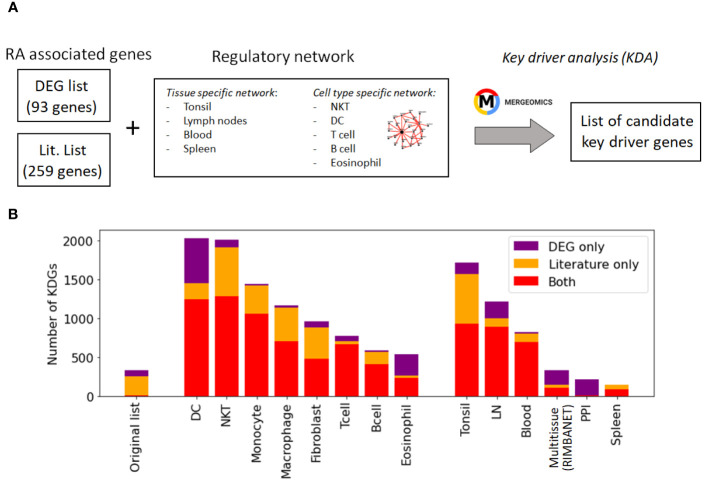
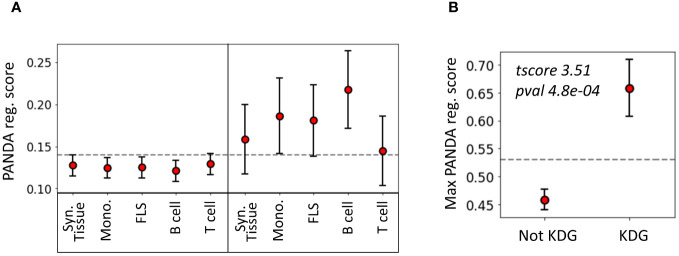
Rheumatoid arthritis (RA) is a common autoimmune and inflammatory disease characterized by inflammation and hyperplasia of the synovial tissues. RA pathogenesis involves multiple cell types, genes, transcription factors (TFs) and networks. Yet, little is known about the TFs, and key drivers and networks regulating cell function and disease at the synovial tissue level, which is the site of disease. In the present study, we used available RNA-seq databases generated from synovial tissues and developed a novel approach to elucidate cell type-specific regulatory networks on synovial tissue genes in RA. We leverage established computational methodologies to infer sample-specific gene regulatory networks and applied statistical methods to compare network properties across phenotypic groups (RA versus osteoarthritis). We developed computational approaches to rank TFs based on their contribution to the observed phenotypic differences between RA and controls across different cell types. We identified 18 (fibroblast-like synoviocyte), 16 (T cells), 19 (B cells) and 11 (monocyte) key regulators in RA synovial tissues. Interestingly, fibroblast-like synoviocyte (FLS) and B cells were driven by multiple independent co-regulatory TF clusters that included MITF, HLX, BACH1 (FLS) and KLF13, FOSB, FOSL1 (B cells). However, monocytes were collectively governed by a single cluster of TF drivers, responsible for the main phenotypic differences between RA and controls, which included RFX5, IRF9, CREB5. Among several cell subset and pathway changes, we also detected reduced presence of Natural killer T (NKT) cells and eosinophils in RA synovial tissues. Overall, our novel approach identified new and previously unsuspected Key driver genes (KDG), TF and networks and should help better understanding individual cell regulation and co-regulatory networks in RA pathogenesis, as well as potentially generate new targets for treatment.
类风湿关节炎(RA)是一种常见的自身免疫性和炎症性疾病,其特征为滑膜组织的炎症和增生。RA 的发病机制涉及多种细胞类型、基因、转录因子(TFs)和网络。然而,人们对 TFs 以及调节滑膜组织中细胞功能和疾病的关键驱动因素和网络知之甚少,而滑膜组织是疾病的发生部位。在本研究中,我们使用来自滑膜组织的可用 RNA-seq 数据库,开发了一种新方法来阐明 RA 滑膜组织基因的细胞类型特异性调节网络。我们利用已建立的计算方法来推断样本特异性基因调控网络,并应用统计方法比较表型组(RA 与骨关节炎)之间的网络特性。我们开发了基于 TFs 对不同细胞类型中 RA 和对照之间观察到的表型差异的贡献来对 TFs 进行排名的计算方法。我们确定了 18 个(成纤维样滑膜细胞)、16 个(T 细胞)、19 个(B 细胞)和 11 个(单核细胞)在 RA 滑膜组织中的关键调节因子。有趣的是,成纤维样滑膜细胞(FLS)和 B 细胞由多个独立的共调节 TF 簇驱动,包括 MITF、HLX、BACH1(FLS)和 KLF13、FOSB、FOSL1(B 细胞)。然而,单核细胞则由一个单一的 TF 驱动簇共同控制,该簇负责 RA 和对照之间的主要表型差异,其中包括 RFX5、IRF9、CREB5。在几个细胞亚群和途径变化中,我们还检测到 RA 滑膜组织中自然杀伤 T(NKT)细胞和嗜酸性粒细胞的存在减少。总体而言,我们的新方法确定了新的和以前未被怀疑的关键驱动基因(KDG)、TF 和网络,这应该有助于更好地理解 RA 发病机制中单个细胞的调节和共调节网络,并可能为治疗提供新的靶点。