Centro Hospitalar e Universitário de São João, Porto, Portugal.
BMC Ophthalmol. 2024 Aug 26;24(1):372. doi: 10.1186/s12886-024-03627-y.
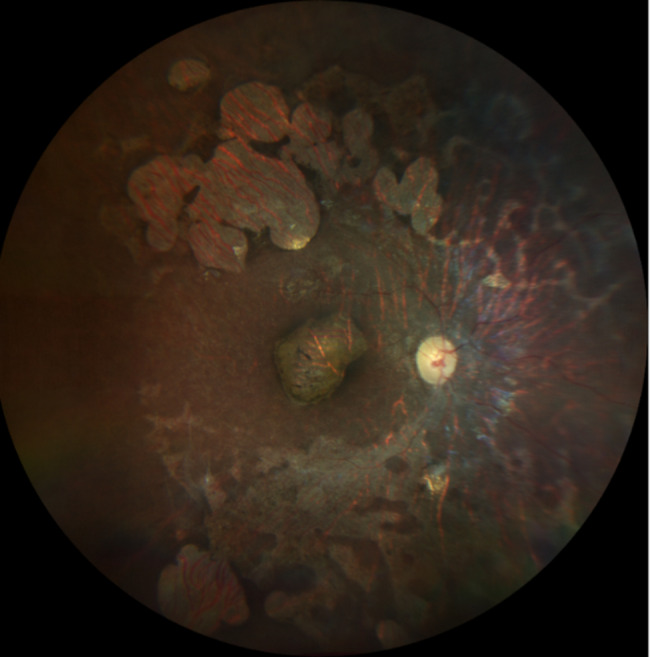
Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation is a rare autosomal dominant disease caused by mutations in KIF11 which disrupt EG5 protein function, impacting the development and maintenance of retinal and lymphatic structures due to its expression in the retinal photoreceptor cilia. The primary ocular finding in MCLMR is chorioretinopathy. Additional features can include microphthalmia, angle-closure glaucoma, persistent hyperplastic primary vitreous, cataract, pseudo-coloboma, persistent hyaloid artery, and myopic or hypermetropic astigmatism. The appearance of the chorioretinal lesions as white to pinkish, round, non-elevated atrophic areas devoid of blood vessels resembles the lacunae in Aicardy syndrome. Due to the lack of systematic description of the lesions and significant phenotypical variability, there is an impending need for a detailed report of each case.
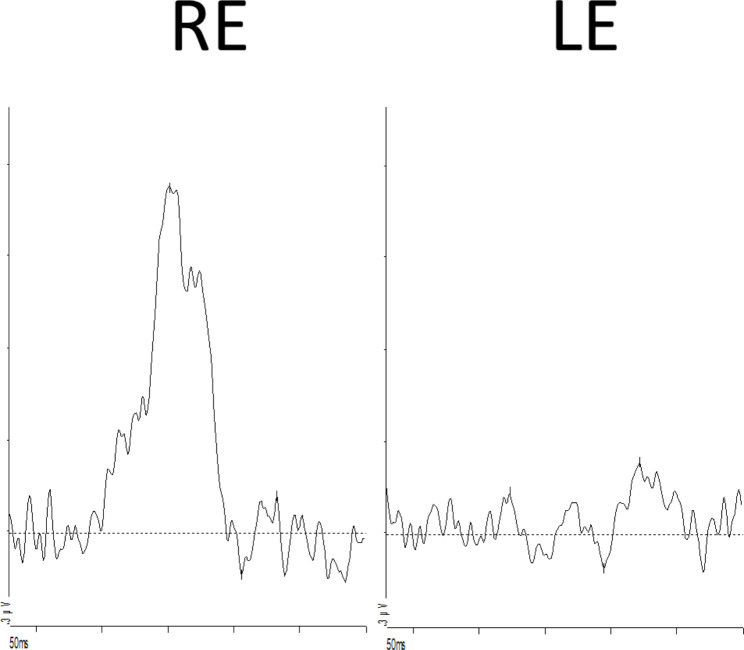
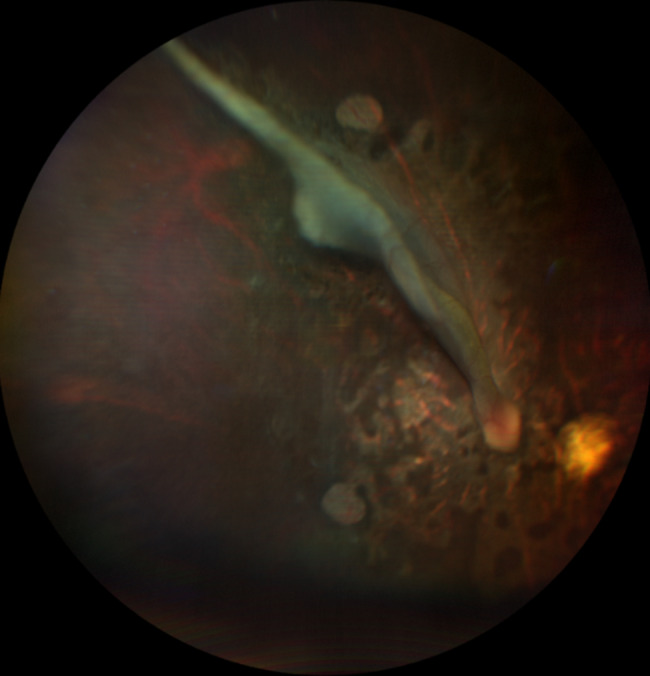
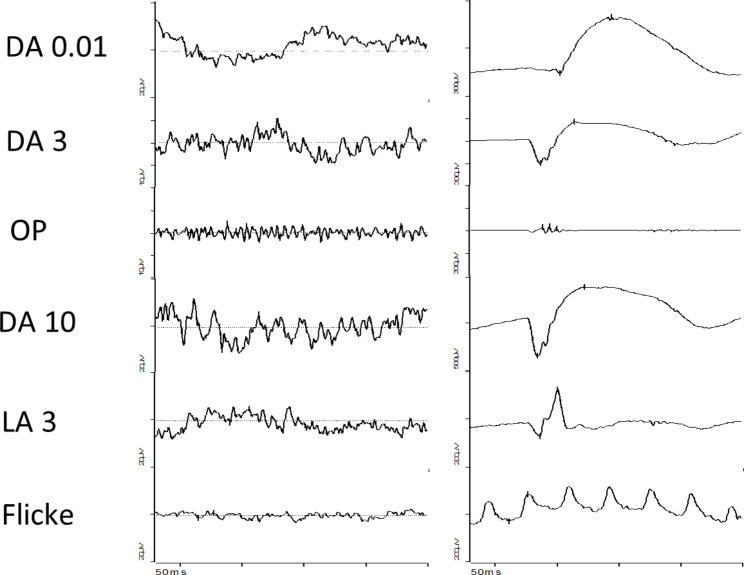
A child with microcephaly detected in the third trimester of gestation began her following in the ophthalmology department due to a non-visually significant cataract. Shortly after, she developed nystagmus and large-angle alternating esotropia with cross-fixation. Her fundus initially showed a pallid optic disc and pigmentary changes, developing thereafter retinal lacunae and a retinal fold. Her differential diagnosis accompanied the dynamic changes in her fundus, which included congenital infections, Leber´s Congenital Amaurosis and Aicardy syndrome. At 19 months old, genetic testing identified a heterozygous mutation (c.1159 C > T, p.Arg387*) in the KIF11 gene. The patient underwent bilateral medial rectus muscle recession surgery at 2 years old for persistent esotropia, with significant improvement. Refraction revealed a hyperopic astigmatism in both eyes (+ 0.25 -2.50 × 180 OD and + 0.75 -2.00 × 170 OS). She continues to require right eye patching for 2 hours daily.
This case report expands the phenotypic spectrum of MCLMR by demonstrating a unique combination of retinal features which sheds new light on differential diagnosis from Aicardy syndrome. Our findings emphasize the significant phenotypic variability associated with MCLMR, particularly regarding ocular involvement. This underscores the importance of detailed clinical evaluation and comprehensive reporting of cases to improve our understanding of the disease spectrum and genotype-phenotype correlations.
微小头畸形伴或不伴脉络膜视网膜病、淋巴水肿或智力迟钝是一种罕见的常染色体显性疾病,由 KIF11 基因突变引起,该突变破坏 EG5 蛋白功能,由于其在视网膜光感受器纤毛中的表达,影响视网膜和淋巴管结构的发育和维持。MCLMR 的主要眼部发现是脉络膜视网膜病。其他特征可能包括小眼球、闭角型青光眼、永存增生性原始玻璃体、白内障、假性裂、永存原始玻璃动脉以及近视或远视散光。脉络膜视网膜病变的外观为白色至粉红色、圆形、无血管的非隆起萎缩区,类似于 Aicardy 综合征中的腔隙。由于缺乏对病变的系统描述和显著的表型变异性,因此迫切需要详细报告每例病例。
一名在妊娠第三个月发现小头畸形的儿童因非视觉显著白内障开始在眼科就诊。不久后,她出现了眼球震颤和大角度交替性外斜视伴交叉固定。她的眼底最初显示苍白的视盘和色素变化,此后发展为视网膜腔和视网膜褶皱。她的鉴别诊断伴随着眼底的动态变化,包括先天性感染、Leber 先天性黑矇和 Aicardy 综合征。19 个月大时,基因检测发现 KIF11 基因的杂合突变(c.1159C>T,p.Arg387*)。2 岁时,患者因持续性外斜视行双侧内直肌后退手术,斜视显著改善。屈光检查显示双眼远视散光(右眼+0.25-2.50×180,左眼+0.75-2.00×170)。她仍需每天右眼遮盖 2 小时。
本病例报告通过展示独特的视网膜特征组合,扩展了 MCLMR 的表型谱,为与 Aicardy 综合征的鉴别诊断提供了新的线索。我们的发现强调了 MCLMR 与显著的表型变异性相关,特别是眼部受累。这突出了详细的临床评估和全面报告病例的重要性,以提高我们对疾病谱和基因型-表型相关性的理解。