Experimental Therapeutics Group, Vall d'Hebron Institute of Oncology (VHIO), Vall d'Hebron Barcelona Hospital Campus, Carrer Natzaret 115-117, 08035, Barcelona, Spain.
Department of Biochemistry and Molecular Biology, Autonomous University of Barcelona, Barcelona, Spain.
Genome Med. 2024 Aug 26;16(1):107. doi: 10.1186/s13073-024-01370-z.
Poly (ADP-ribose) polymerase 1 and 2 (PARP1/2) inhibitors (PARPi) are targeted therapies approved for homologous recombination repair (HRR)-deficient breast, ovarian, pancreatic, and prostate cancers. Since inhibition of PARP1 is sufficient to cause synthetic lethality in tumors with homologous recombination deficiency (HRD), PARP1 selective inhibitors such as saruparib (AZD5305) are being developed. It is expected that selective PARP1 inhibition leads to a safer profile that facilitates its combination with other DNA damage repair inhibitors. Here, we aimed to characterize the antitumor activity of AZD5305 in patient-derived preclinical models compared to the first-generation PARP1/2 inhibitor olaparib and to identify mechanisms of resistance.
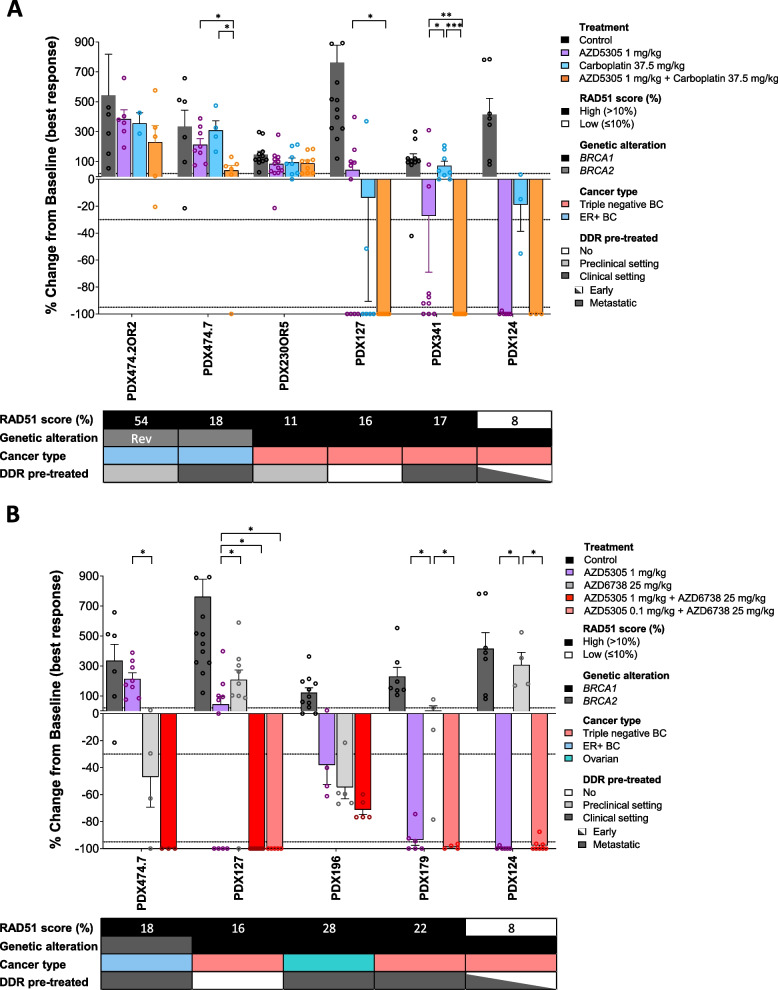
Thirteen previously characterized patient-derived tumor xenograft (PDX) models from breast, ovarian, and pancreatic cancer patients harboring germline pathogenic alterations in BRCA1, BRCA2, or PALB2 were used to evaluate the efficacy of AZD5305 alone or in combination with carboplatin or an ataxia telangiectasia and Rad3 related (ATR) inhibitor (ceralasertib) and compared it to the first-generation PARPi olaparib. We performed DNA and RNA sequencing as well as protein-based assays to identify mechanisms of acquired resistance to either PARPi.
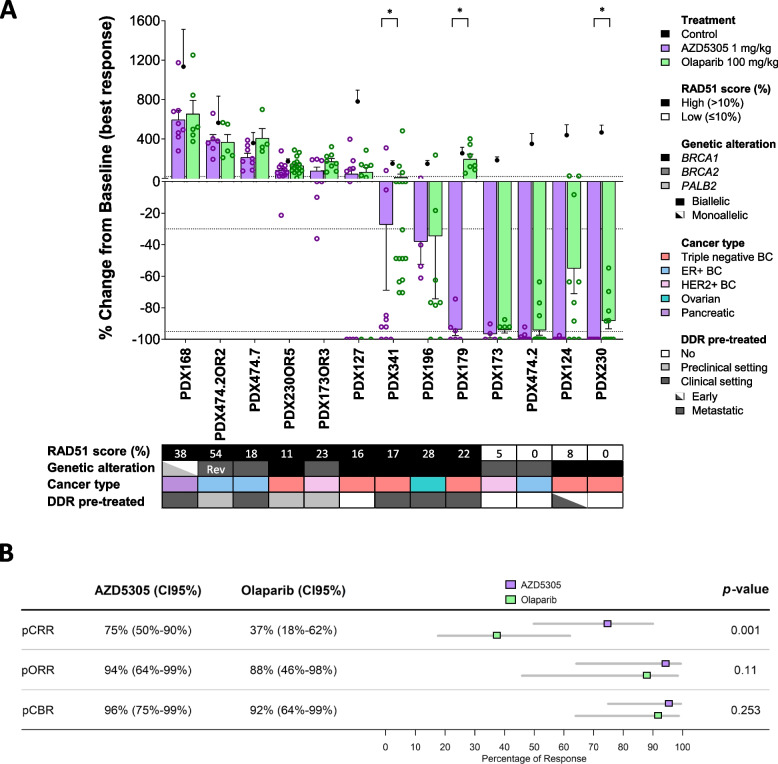
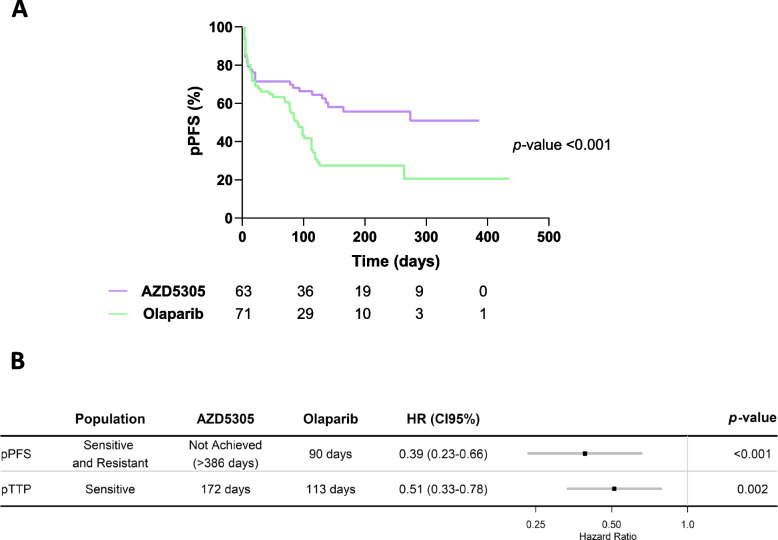
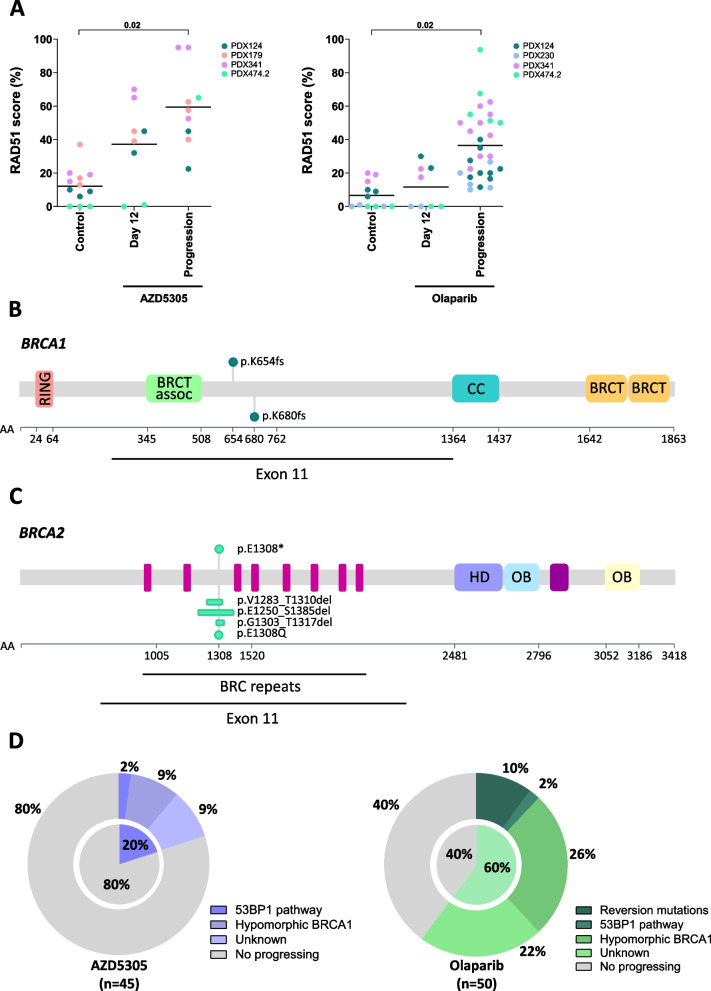
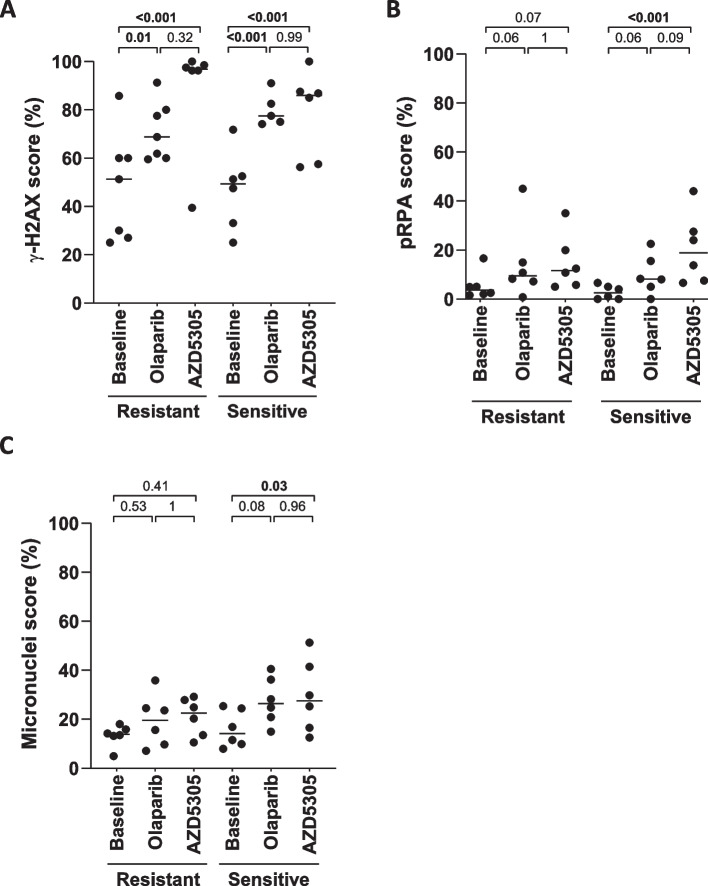
AZD5305 showed superior antitumor activity than the first-generation PARPi in terms of preclinical complete response rate (75% vs. 37%). The median preclinical progression-free survival was significantly longer in the AZD5305-treated group compared to the olaparib-treated group (> 386 days vs. 90 days). Mechanistically, AZD5305 induced more replication stress and genomic instability than the PARP1/2 inhibitor olaparib in PARPi-sensitive tumors. All tumors at progression with either PARPi (39/39) showed increase of HRR functionality by RAD51 foci formation. The most prevalent resistance mechanisms identified were the acquisition of reversion mutations in BRCA1/BRCA2 and the accumulation of hypomorphic BRCA1. AZD5305 did not sensitize PDXs with acquired resistance to olaparib but elicited profound and durable responses when combined with carboplatin or ceralasertib in 3/6 and 5/5 models, respectively.
Collectively, these results show that the novel PARP1 selective inhibitor AZD5305 yields a potent antitumor response in PDX models with HRD and delays PARPi resistance alone or in combination with carboplatin or ceralasertib, which supports its use in the clinic as a new therapeutic option.
聚(ADP-核糖)聚合酶 1 和 2(PARP1/2)抑制剂(PARPi)是获批用于同源重组修复(HRR)缺陷型乳腺癌、卵巢癌、胰腺癌和前列腺癌的靶向治疗药物。由于抑制 PARP1 足以导致同源重组缺陷(HRD)的肿瘤发生合成致死,因此正在开发 PARP1 选择性抑制剂,如鲁卡帕尼(AZD5305)。预计选择性 PARP1 抑制会产生更安全的特征,从而促进其与其他 DNA 损伤修复抑制剂的联合应用。在这里,我们旨在比较 AZD5305 与第一代 PARP1/2 抑制剂奥拉帕利在患者来源的临床前模型中的抗肿瘤活性,并确定耐药机制。
使用 13 个先前经过特征鉴定的来自乳腺癌、卵巢癌和胰腺癌患者的肿瘤异种移植(PDX)模型,这些患者携带 BRCA1、BRCA2 或 PALB2 种系致病性改变,以评估 AZD5305 单独或联合卡铂或共济失调毛细血管扩张症和 Rad3 相关(ATR)抑制剂(塞拉西替布)的疗效,并与第一代 PARPi 奥拉帕利进行比较。我们进行了 DNA 和 RNA 测序以及基于蛋白质的测定,以鉴定对任何 PARPi 获得性耐药的机制。
AZD5305 在临床前完全缓解率(75%对 37%)方面显示出比第一代 PARPi 更好的抗肿瘤活性。与奥拉帕利治疗组相比,AZD5305 治疗组的临床前无进展生存期显著延长(>386 天对 90 天)。在 PARPi 敏感的肿瘤中,AZD5305 比 PARP1/2 抑制剂奥拉帕利诱导更多的复制应激和基因组不稳定性。所有用 PARPi 进展的肿瘤(39/39)均通过 RAD51 焦点形成显示出 HRR 功能的增加。鉴定出的最常见的耐药机制是 BRCA1/BRCA2 中获得的回复突变和 BRCA1 的低功能积累。AZD5305 不能使对奥拉帕利获得性耐药的 PDX 对其敏感,但与卡铂或塞拉西替布联合使用时,在 3/6 和 5/5 个模型中分别产生了深刻和持久的反应。
总的来说,这些结果表明,新型 PARP1 选择性抑制剂 AZD5305 在 HRD 的 PDX 模型中产生了强大的抗肿瘤反应,并延迟了 PARPi 单独或与卡铂或塞拉西替布联合使用时的耐药性,这支持其在临床上作为一种新的治疗选择。